Table of Contents
Amyotrophic Lateral Sclerosis (ALS)
Figure 1. Motor Neuron.
What is Amyotrophic Lateral Sclerosis (ALS)?
Amyotrophic Lateral Sclerosis (ALS) is also known as Lou Gehrig’s disease, or motor neuron disease. It is characterized by the gradual paralysis of people as the brain is no longer able to communicate with the muscles of the body. It is a type of progressive disease, meaning it gets worse over time. People with this disease eventually lose the ability to walk, talk, eat, swallow, and even breathe as the muscles of the body break down. There is no known cure and there are few treatment options available (“What is ALS? What Are the Types and Causes?,” 2016).
Two Types of ALS
Sporadic ALS
When the disease occurs spontaneously, affecting anyone regardless of their gender, ethnicity or age (“What is ALS? - ALS Society of Canada,” n.d.). This is the most common form of ALS, presenting in approximately 95% of ALS cases (“What is ALS? What Are the Types and Causes?,” 2016).
Familial ALS
When the onset of the disease is due to genetics, in other words is passed on from parent to child. This type of the disease occurs in approximately 5 to 10% of ALS cases (“What is ALS? - ALS Society of Canada,” n.d.).
Signs and Symptoms
- Reduced strength in the arms and/or legs
- Difficulty speaking
- Difficulty swallowing
- Difficulty breathing
- Muscles begin to atrophy
- Paralysis
(“About - Project MinE,” n.d.)
Statistics
Prevalence: ALS most often affects people between the ages of 40 and 60. More than 200,000 people around the world are living with ALS, and of that an estimated 3,000 Canadians.
Mortality Rate: In Canada, approximately 2-3 people die of ALS per day.
Incidence: A little over 5,000 people in the U.S. are diagnosed with ALS each year, which translates to about 15 new cases a day (“Facts You Should Know,” n.d.).
Prognosis: 80% of people with ALS die within 2-5 years of diagnosis.
(“What is ALS? - ALS Society of Canada,” n.d.)

Figure 2. ALS patient in hospital.
Etiology
There is no clear answer for what causes ALS; however, studies have shown that both genetics and the environment plays a role in acquiring the disease.
Genetics
Genetics impacts the development of ALS due to many various factors. Firstly various gene mutations can cause ALS. Scientists discovered that gene mutations in the SOD1 genes were connected to familial ALS as they had the ability to degenerate motor neurons (Mayo Clinic, 2018). ALS can also be due to chemical imbalances. People with ALS usually have high levels of glutamate, a chemical messenger in the brain, which is toxic to neurons (Mayo Clinic, 2018). Lastly, ALS can be due to an altered immune system in which the immune system attacks nerve cells, leading to its death (Mayo Clinic, 2018).
Environment
ALS can also be caused by the environment (Mayo Clinic, 2018). Kamel et al. (2005) believe that exposure to toxins or infectious agents cause ALS – especially in those who are exposed to high levels of lead (Kamel et al., 2005). Secondly, studies show that physical traumas and previous hospitalization are correlated to a higher risk of ALS. Specific studies also show that physical traumas at earlier ages leads to a higher risk in ALS (Seals, Hansen, Gredal, & Weisskopf, 2016). Lastly, a study by Swash (2018) has shown that increased physical activity has a linear association with ALS as it is linked to excitotoxicity, or nerve cell death of the central nervous system (Swash, 2018).
Pathogenesis
Familial ALS and Sporadic ALS are two forms of ALS, which can be distinguished by diagnostic criteria and their pathophysiology. The origin of ALS is multifactorial as the onset of the disease can be due to many disturbances in the molecular pathways. The most familiar pathway known to causes ALS is through a mutation of the antioxidant enzyme known as superoxide dismutase 1 (SOD1) (Zariei et al., 2015). A mutation in the gene that encodes for the SOD1 enzyme leads to the growth of mutant SOD1 which has a very unstable structure (Zariei et al., 2015). This unstable structure leads to misfolding in the Mutant SOD1 which initiates aggregation in the central nervous systems motor neurons (Zariei et al., 2015). There are many mechanisms and pathways Mutant SOD1 can take that lead to neurodegeneration. Some of the main hypotheses that have been researched and observed in ALS patients are free radical-mediated oxidative stress, functional abnormalities of mitochondria, glutamate excitotoxicity, and impairment of axonal structure or transport defects (Zariei et al., 2015).
Free radical-mediated oxidative stress
SOD1 gene plays a major role in the maintenance of cells. The SOD1 enzyme functions to bind zinc and copper molecules in order to breakdown free radicals and toxic molecules, which will prevent cell damage (SOD1, 2016). The mutation in the SOD1 gene seen in ALS patients is shown to have a cytotoxic effect. Mutant SOD1 loses its function to bind zinc and copper together to eliminate the free radicals or other toxins. This can lead to oxidative stress since there is an accumulation of free radicals (Zariei et al., 2015; Kiernan et al., 2011).
Functional Abnormalities of Mitochondria
Mitochondria play a major role in the cell as they create ATP energy using the electron transport chain. Mitochondria dysfunctionality is another major problem seen in ALS patients (Zariei et al., 2015). The mutant SOD1 accumulates in the intermembrane space and the matrix of the mitochondria, which leads to abnormalities (Zariei et al., 2015; Fischer et al., 2010). Abnormalities include ATP deficits, which can lead to neurodegeneration, as there is a lack of energy required for the cell to function efficiently (Zariei et al., 2015). More specifically, researchers have noticed that the accumulation causes a decrease in the activity of complex I and IV of the electron transport chain. Complex I is in charge of accepting the electron from NADPH, and complex IV is the final complex that the electron passes to before being accepted by oxygen and becoming H20 (Learning., n.d.). With the decrease in activity of these major complexes, there is a decrease in electrons moving across the ETC, binding to oxygen, and creating a gradient, which overall translates to a decrease in ATP production. In addition, mitochondria play a role in apoptosis for certain proteins and the aggregated mutant SOD1 hinders this process. B-cell lymphoma 2 (Bcl-2) is the protein that regulates cell death for mitochondrial-dependent apoptosis (Zariei et al., 2015). The aggregated SOD1 leads to stimulation of Bcl-2. This can cause premature death of these cells, leading to neuronal dysfunction and neuromuscular degeneration (Zariei et al., 2015).
Glutamate Excitotoxicity
Glutamate is a neurotransmitter that plays a major role in the neurotransmission process. Glutamate is made in the presynaptic terminal and released into the synaptic cleft (Zariei et al., 2015). It travels through the cleft to activate the postsynaptic receptors. Once the postsynaptic receptors have been activated, the glutamate neurotransmitter is removed using the excitatory amino acid transporters (EAATs) (Zariei et al., 2015). The picture on the right side displays the maintenance of glutamate molecules through the use of healthy EAATs. The picture on the left shows the absences of EAATs in an individual with ALS and the accumulation of Glutamate. This continuous cycle of release and elimination of glutamate is important in maintaining a gradient and avoiding excitotoxic neuronal damage (Zariei et al., 2015). Researchers have observed a decrease in function of the EEATs in ALS patients and transgenic SOD1 mouse models (Zariei et al., 2015). This is extremely harmful as it increases the amount of glutamate present in the synaptic cleft, which causes over-stimulation of the glutamate receptors. The over-stimulation leads to excitotoxicity and neuronal degeneration (Zariei et al., 2015).
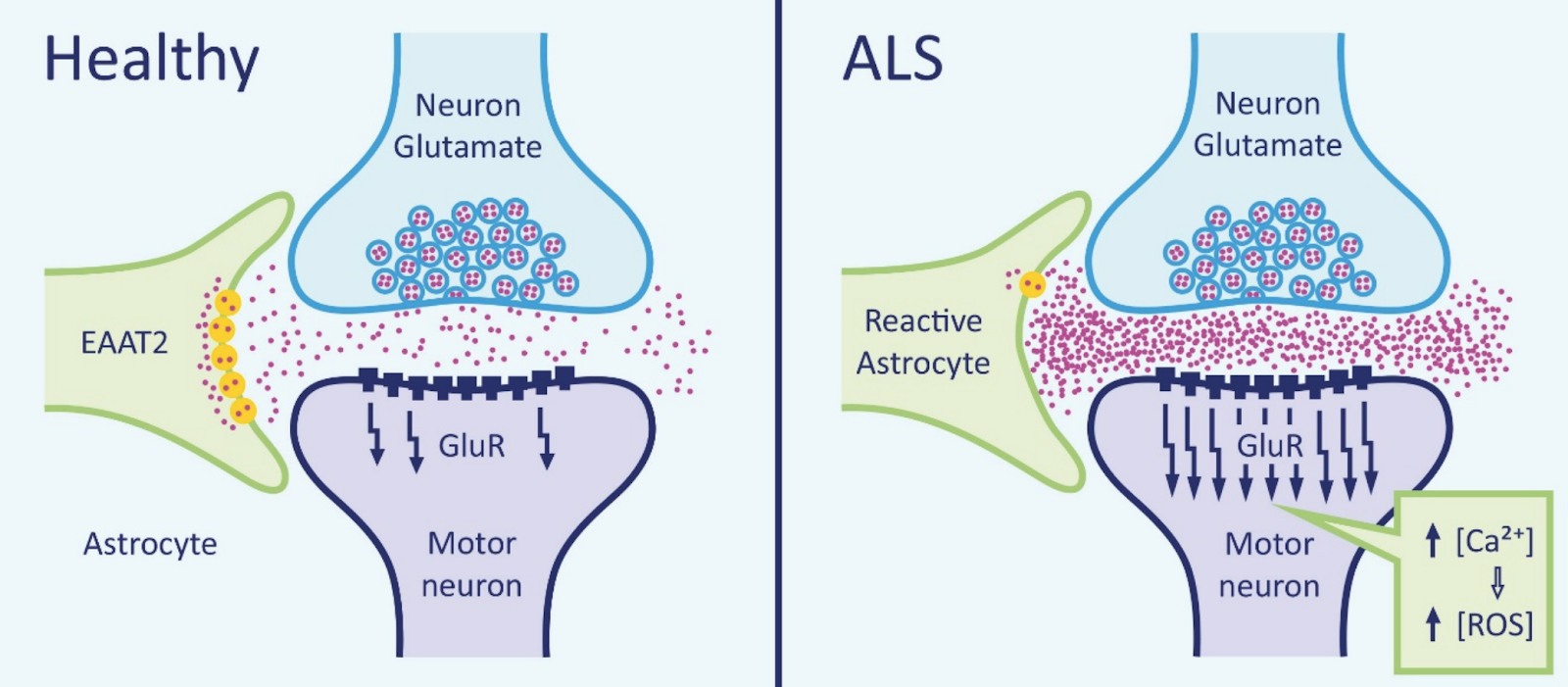
Figure 3. Mechanism of glutamate excitotoxicity.
Impairment of Axonal Structure or Transport Defects
Impairment of axonal transport and structure can be caused by many different pathways triggered by mutant SOD1 (Zariei et al., 2015). Mechanisms such as the lack of mitochondrial function, disruption of kinesin function, energy depletion, and excitotoxicity caused by glutamate, lead to the impairment of axonal transport/structure (Zariei et al., 2015). Many experiments conducted on mice with mutant SOD1 displayed loss of defects in axonal transport in the early process of the disease. The impairment of the structure leads to build up of mitochondria, autophagosomes, and neurofilaments in the damaged motor neurons, which eventually causes the death of the neurons (Zariei et al., 2015).
Diagnosis
Criteria
There is no distinct biological marker that is available for study in the diagnosis of ALS. Due to this, various medical and laboratory tests need to be completed to rule out other diseases that exhibit identical or similar symptoms (e.g. electrophysiological studies, neuroimaging studies, nerve conduction studies, etc.). The El Escorial criteria was typically used as the international standard of guidelines for diagnosing ALS. However, a conference held in April 1998 in Warrenton, Virginia by the World Federation of Neurology Research Committee on Motor Neuron Diseases. The figure below is an excerpt of the consensus document that was produced at the conference stating the following requirements for proper ALS diagnosis (Brooks et al., 2000).

Figure 4. Requirements for ALS Diagnosis.
Renamed as the Airlie House criteria in 1998 after the El Escorial was revised, these set of guidelines went under further revision to produce Awaji-Shima criteria. This was introduced in 2008, which requires clinical or electrophysiological evidence with more regional specifications (e.g. signs in bulbar, spinal regions etc.) to ensure that an accurate ALS diagnosis is being made. The table below compares the original 1994 El Escorial criteria with the revised 1998 Airlie House (incorporated with the 2008 Awaji-Shima criteria) to demonstrate how there has been an increase in the specificity of diagnosing ALS. (Source: Hardiman et al., 2011)
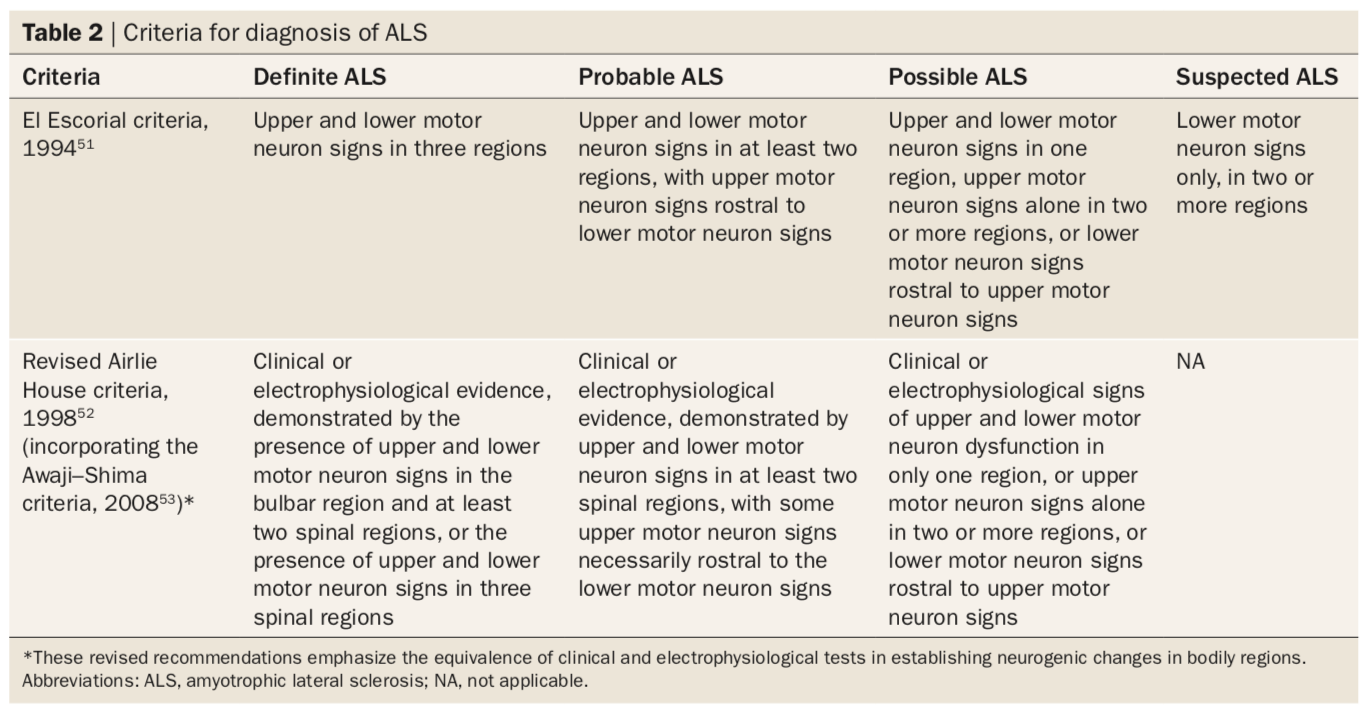
Figure 5. Criteria for diagnosis of ALS.
Potential ALS Biomarkers in Research
Seeing as there is no unique biomarker to ALS and its cause is undetermined, a 2000 study by Bogdanov et al. discusses how damage to neurons could be attributed to oxidation. A biomarker in cerebrospinal fluid, urine, and plasma that indicates DNA damage is 8-hydroxy-2’-deoxyguanosine (8OH2’dG). The study, involving 165 subjects, examined this marker in ALS patients against a control group for almost a year. The results indicated that as the neurodegenerative properties worsened in ALS subjects over the course of the study, the levels of 8OH2’dG increased. These results are coherent with the hypothesis that “oxidative pathology accompanies…ALS and [suggests] that 8OH2’dG may provide a useful tool for monitoring therapeutic interventions” (Bogdanov et al., 2000) across the progression of the disease.
A 2018 study by Gonzalez-Garza and colleagues studied adipsin, MIP-1b, and IL-8 as potential biomarkers for diagnosis of ALS. ALS could be an autoimmune disease as there is evidence that an immune-mediated inflammatory process parallels the progression of ALS (Gonzalez-Garza et al., 2018). In their study, the researchers “analyzed 19 different cytokines and growth factors in the cerebrospinal fluid of 77 ALS patients and 13 controls…” Their results showed an increase of adipsin, MIP-1b, and IL-6, and a decrease of IL-8 thresholds, related with ALS patients” Gonzalez-Garza et al., 2018). Furthermore, their study’s conclusion was found using techniques classification tree analysis and PanelomiX algorithm analysis.
Tests and Examinations
No single test can give an explicit ALS diagnosis. Neurological tests can assist in an initial visual-based diagnosis of ALS; however, further examinations, such as EMG, blood and urine tests, and muscle and nerve biopsies can be used to confirm or refute the diagnosis (Mitsumoto, 1997).
Electromyography (EMG): This is a helpful test in the beginning stages of ALS as it detects lower motor neuron degeneration in specific regions to rule out other potential illnesses. Furthermore, EMGs are especially useful in ruling out ALS symptoms involving the upper motor neuron – this dismisses diseases such as multiple sclerosis and cervical spondylitic myelopathy. EMGs involve nerve conduction studies and examinations that use needle electrodes on the tongue and lower face. To support a diagnosis of ALS, EMG tests measure a regular or decreased amplitude of compound motor action potentials and other factors (e.g. chronic neurogenic motor units, etc.) (Mitsumoto, 1997).
Neuroimaging: Tests such as radiography, myelography, CT scans, and MRIs can also be used to determine whether an ALS diagnosis can be supported. However, in recent times, MRIs have effectively been deemed to replace the role which was played by myelography and CT scans. MRIs are especially useful in recognizing whether any tumours or lesions can cause upper motor neuron symptoms.
Muscle and Nerve Biopsy: This is an optional test performed for the “differential diagnosis of ALS”. Particular cases of inclusion body myositis appear nearly identical to the lower motor neuron symptoms of ALS. As such, biopsies may be performed to rule out such conditions.
Laboratory Tests: Blood and urine tests are taken for blood counts (CBCs), and measures of certain electrolytes and proteins (antibodies). These tests can rule out conditions, such as hyperparathyroidism and myasthenia.
Treatment
Riluzole
Riluzole is the main drug that is utilized for treating ALS. Riluzole specifically works in blocking the release of glutamate from one neuron to the next as shown in the figure below (Rothstein, 1996). Glutamate is the neurotransmitter that is used for majority of the excitatory functions in the brain. Many of the functions of glutamate include increase cell membrane permeability, and activating cell surface receptors (Stephen et al., 2010). However, when there is an excessive release of glutamate it can result in excitotoxicity due to the large influx of calcium in the the neuron (Lewerenz & Maher, 2015). Motor neurons specifically are more sensitive to calcium release, as a result, causing neuron death. Hence, Riluzole inhibits the release of glutamate from the presynaptic neuron, stopping the influx of calcium into the neuron (Rothstein, 1996). Riluzole is not a cure to ALS, however, the drug can help prolong life by delaying the onset of muscle weakness and paralysis. This would also include the reduction of the reliance of patients to tracheostomy, which is a procedure in which an incision is created in a patients neck to place a tube in, as an alternative method of breathing (Bryson, Fulton & Benfield, 1996).

Figure 6. The mechanism of Riluzole.
Stem Cells
One potential treatment that is currently being studied in mice is associated with stem cell therapy. Kondo et al. (2014), transplanted glial-rich neural progenitors from human induced pluripotent cells into the lumbar spinal cord of mice with ALS. These diseased mice had mutant superoxide dismutase 1 (SOD1) enzymes that promotes ALS development and the loss of function in motor neurons. After transplantation with hiPSCs, the cells were able to differentiate into astrocytes resulting in an increased life span. Researchers further explains that this therapy is beneficial because the AKT signaling pathway in astrocytes is activated which promotes the survival of cells in ALS. This is because it replaces mutated ALS astrocytes, which originally mediates the accumulation of glutamate and ultimately cell death (Pehar, Harlan, Killoy, & Vargas, 2018). Kondo et al. (2014), explains that although this experiment only shows an improvement in the motor abilities of the lower limb, there is a possibility of targeting different areas of the spinal cord to treat different symptoms (Kondo et al., 2014).

Figure 7. Article exploring the use of stem cells in ALS treatment.
CuATSM
Treatment with a drug called diacetylbis(N(4)-methylthiosemicarbazonato) copper II (CuATSM) has the potential to reduce the symptoms, and slow the advancement of familial ALS. The role of this drug is to penetrate the blood-brain barrier and deliver copper to body cells that are copper deficient (Irving, 2019). According to ALS Therapy Development Institute (2016), this is significant as these cells are able to use copper to prevent the misfolding of proteins that causes ALS (ALS Therapy Development Institute, 2016). According to Xia (n.d.), In humans it is specifically to prevent the misfolding of the SOD1 protein which would affect motor neurons by causing endoplasmic reticulum, proteasome, and mitochondrial stress that influences cell death (Xia, n.d.). Furthermore, mutated SOD1 modifies many cellular pathways and functions including axonal transport, RNA processing, etc. which leads to muscle atrophy (Xia, n.d.). Carvalho (2019), concluded that CuATSM improves lung and cognitive function during Phase 1 human clinical trials (Carvalho, 2019). This was measured using the forced vital capacity and Edinburgh cognitive and behavioural ALS Screen (ECAS) scoring system (Carvalho, 2019). Patients with lower doses of CuATSM experienced minor improvements in these scores over 24 weeks compared to those with higher doses (Carvalho, 2019). Phase 2 clinical trials will be held in 2019 to confirm its impact on biological activities using larger and randomized sample sizes, implementing placebo controls, and using a double-blind experiment (Irving, 2019).

Figure 8. Structure of CuATSM.
ALS PowerPoint
References
About - Project MinE. (n.d.). Retrieved from https://www.projectmine.com/about/
Amyotrophic lateral sclerosis (ALS). (2018). Retrieved from https://www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms- causes/syc-20354022
Amyotrophic Lateral Sclerosis (ALS): Facts & Symptoms of Lou Gehrig's Disease. (22, June). Retrieved from https://www.livescience.com/39583-als-lou-gehrigs-disease.html
Amyotrophic Lateral Sclerosis (ALS) Fact Sheet | National Institute of Neurological Disorders and Stroke. (2018). Retrieved from https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet
Amyotrophic lateral sclerosis (ALS) - Symptoms and causes. (2018). Retrieved from https://www.mayoclinic.org/diseases-conditions/amyotrophic- lateral-sclerosis/symptoms-causes/syc-20354022
Bogdanov, M., Brown Jr, R. H., Matson, W., Smart, R., Hayden, D., O’Donnell, H., … & Cudkowicz, M. (2000). Increased oxidative damage to DNA in ALS patients. Free Radical Biology and Medicine, 29(7), 652-658.
Bryson, H. M., Fulton, B., & Benfield, P. (1996). Riluzole. Drugs, 52(4), 549–563. https://doi.org/10.2165/00003495-199652040-00010
Brooks, B. R., Miller, R. G., Swash, M., & Munsat, T. L. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders, 1(5), 293-299.
Carvalho, J. (2019). ALS Progression May Be Slowed With CuATSM Treatment, Trial Shows. Retrieved from https://alsnewstoday.com/2019/01/10/cuatsm- may-slow-als-progression-improve-cognition-respiration-phase-1-trial/
Copper Compound (CuATSM) Shows Promise in ALS Lab Studies. (n.d.). Retrieved from https://www.als.net/news/copper-compound-cuatsm-shows-promise-in- als-laboratory-results/
Costa, J., Swash, M., & de Carvalho, M. (2012). Awaji criteria for the diagnosis of amyotrophic lateral sclerosis: a systematic review. Archives of neurology, 69(11), 1410-1416.
Doble, A. (1996). The pharmacology and mechanism of action of riluzole. Neurology, 47(6 Suppl 4), S233-241.
Facts You Should Know. (n.d.). Retrieved from http://www.alsa.org/about-als/facts-you-should-know.html
Fischer, L. R., Igoudjil, A., Magrane, J., Li, Y., Hansen, J. M., Manfredi, G., & Glass, J. D. (2010). SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain, 134(1), 196-209.
Gonzalez-Garza, M. T., Martinez, H. R., Cruz-Vega, D. E., Hernandez-Torre, M., & Moreno-Cuevas, J. E. (2018). Adipsin, MIP-1b, and IL-8 as CSF Biomarker Panels for ALS Diagnosis. Disease markers, 2018.
Hardiman, O., Van Den Berg, L. H., & Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nature reviews neurology, 7(11), 639.
Irving, M. (2019). New ALS drug slows disease progression in groundbreaking clinical trial. Retrieved from https://newatlas.com/als-mnd-drug- clinical-trial-success/58026/?fbclid=IwAR2b0PRNHR7mmPCO0UVCq6KSHvwAQonjQtZgvQDgsbUrEC2Unrw7k0qROzU
Kamel, F., Umbach, D., Hu, H., Munsat, T., Shefner, J., Taylor, J., & Sandler, D. (2005). Lead Exposure as a Risk Factor for Amyotrophic Lateral Sclerosis. Neurodegenerative Diseases,2(3-4), 195-201. doi:10.1159/000089625 (https://www.ncbi.nlm.nih.gov/pubmed/16909025)
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., … & Zoing, M. C. (2011). Amyotrophic lateral sclerosis. The lancet, 377(9769), 942-955.
Kondo, T., Funayama, M., Tsukita, K., Hotta, A., Yasuda, A., Nori, S., . . . Inoue, H. (2014). Focal Transplantation of Human iPSC-Derived Glial- Rich Neural Progenitors Improves Lifespan of ALS Mice. Stem Cell Reports,3(2), 242-249. doi:10.1016/j.stemcr.2014.05.017(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4175543/pdf/main.pdf)
Learning, L. (n.d.). Biology for Majors I. Retrieved from https://courses.lumenlearning.com/wm-biology1/chapter/reading- electron-transport-chain/
Lewerenz, J., & Maher, P. (2015). Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Frontiers in Neuroscience, 9. https://doi.org/10.3389/fnins.2015.00469
Mitsumoto, H. (1997). Diagnosis and progression of ALS. Neurology, 48(4 Suppl 4), 2S-8S.
Pehar, M., Harlan, B. A., Killoy, K. M., & Vargas, M. R. (2018). Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Current Pharmaceutical Design,23(33). doi:10.2174/1381612823666170622095802
Riluzole Oral : Uses, Side Effects, Interactions, Pictures, Warnings & Dosing - WebMD. (n.d.). Retrieved from https://www.webmd.com/drugs/2/drug-12138/riluzole-oral/details
Rothstein, J. D. (1996). Therapeutic horizons for amyotrophic lateral sclerosis. Current Opinion in Neurobiology, 6(5), 679–687. https://doi.org/10.1016/S0959-4388(96)80103-6
Seals, R. M., Hansen, J., Gredal, O., & Weisskopf, M. G. (2016). Physical Trauma and Amyotrophic Lateral Sclerosis: A Population-Based Study Using Danish National Registries. American Journal of Epidemiology,183(4), 294-301. doi:10.1093/aje/kwv169 (https://academic.oup.com/aje/article/183/4/294/2195444)
SOD1 gene - Genetics Home Reference - NIH. (2016). Retrieved from https://ghr.nlm.nih.gov/gene/SOD1
Swash, M. (2018). Physical activity as a risk factor in ALS. Journal of Neurology, Neurosurgery & Psychiatry,89(8), 793-793. doi:10.1136/jnnp-2018-318147
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., … Dingledine, R. (2010). Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacological Reviews, 62(3), 405–496. https://doi.org/10.1124/pr.109.002451
What is ALS? - ALS Society of Canada. (n.d.). Retrieved from https://www.als.ca/about-als/what-is-als/
What is ALS? What Are the Types and Causes? (2016). Retrieved from https://www.webmd.com/brain/understanding-als-basics#1
Xia, M. (n.d.). Investigating the role of PICK1 in regulating mutant SOD1 in amyotrophic lateral sclerosis. Nature Education, 8(3),4. doi:10.14711/thesis-991012588267103412
Zarei, S., Carr, K., Reiley, L., Diaz, K., Guerra, O., Altamirano, P. F., . . . Chinea, A. (2015). Surgical Neurology International, 6, 171st ser. doi:https://dx.doi.org/10.4103/2152-7806.169561