Table of Contents
Cystic Fibrosis
In 1938, cystic fibrosis was first recognized as a separate disease from celiac disease (Anderson, 1938). Anderson’s clinical and pathological study revealed mucus plugging of the glandular ducts of the pancreas in malnourished infants (Anderson, 1938). Cystic fibrosis is also known as “mucoviscidosis” due to the ducts being clogged by the mucus (Farber, 1944). A clue of the causation for this disease was discovered in the defect of sweat electrolyte concentration which was consistent in cystic fibrosis patients (DI Sant'Agnese et al., 1953). Chloride transport protein was identified as the physiological defect for this disease (Quinton, 1983; Knowles et al., 1983). These discoveries allowed accurate methods of diagnostic testing such as the sweat test and improved treatment such as the pillars of care (Davis, 2006).
Epidemiology
Cystic fibrosis affects approximately 70,000 people worldwide and 30,000 people in North America and Europe (Grief, 2008). Usually cystic fibrosis have an incidence of 1 in 2,500 however it varies depending on region (Cystic Fibrosis Canada, 2012). Incidence is how many new cases there are for every 100 000 people. Most of these new incidence cases occur in newborns (Hodson, 2007). Caucasians has the highest birth prevalence (number of people with cystic fibrosis per 10,000 live births) 1 in 2,500, compared to any other racial group. Asians are less likely to have cystic fibrosis with a birth prevalence of 1 in 35,000 (Grosse,et al., 2004). Both male and female newborns are equally likely to have cystic fibrosis (Grosse,et al., 2004).
In 1934 it was thought that cystic fibrosis is fatal in infancy (Preedy, 2012). With improved nutrition, treatment and diagnostic tests 80% of patients will reach adulthood (Preedy, 2012). In 2006 it was predicted that the median survival age was 36.5 years (Davis, 2006). Now, the latest predicted median age of survival for Canadians with cystic fibrosis is 50 years old. (Cystic Fibrosis Canada, 2012). More than 35% of patients with cystic fibrosis are older than 18 (Davis, 2006). As adults most of them finish school, join the workforce, pay taxes, marry, start families, and generally take on the problems of everyday life (Cystic Fibrosis Foundation, 2012). However, hospitalization and home therapy become more frequent and extensive as the patients age and the disease advance (Davis, 2006). Annual death rates in cystic fibrosis patients are less than 2% (Mall & Elborn, 2014). The most common cause of death in patients with cystic fibrosis is chronic lung infection (Mall & Elborn, 2014).
Signs and symptoms
Individuals with cystic fibrosis experiences many signs and symptoms that involves multiple systems in the body (Cystic Fibrosis Canada, 2012). The severity of the symptoms varies from person to person depending the presence or function of the CFTR protein (Davis, 2006).
Integumentary System
- Salty-tasting skin
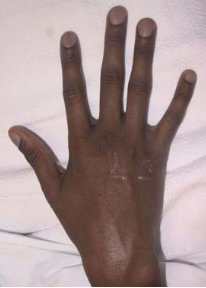
- Digital clubbing.
The skin taste like salt because two to five times the normal amount of salt in sweat (Seeley & Tate, 2008). This can induce rapid dehydration (Seeley & Tate, 2008). Clubbing is an enlargement of the finger and toe because of proliferation of the connective tissue (Davis 2006; Seeley & Tate, 2008).
A 16-year-old boy with cystic fibrosis showing club fingers (Chen, 2006).
Respiratory System
- Consist of recurrent or persistent chest infections
- Coughing with phlegm
- Wheezing or shortness of breath
These symptoms arise because of the mucus buildup in the lungs is a perfect habitat for bacteria (O'Sullivan & Freedman, 2009). As this disease progress throughout a patient’s life, lung bleeding or collapsed lung may occur (Seeley & Tate, 2008). Also, severe lung disease may result an enlargement of the right ventricle of the heart. The heart's right ventricle enlarge to increase the force needed to pump the blood to the lungs (Seeley & Tate, 2008).
A child with cystic fibrosis coughing (Cystic Fibrosis Canada, 2012)

Digestive System
- Poor growth
- Weight loss
- Abnormal bowel movement
- Smelly greasy stools.
The mucus buildup blocks of ducts of important enzyme secreting organs such as the pancreas and liver (Seeley & Tate, 2008). The blockage of pancreatic ducts disallows enzymes to break down the nutrients in the food to be absorbed (O'Sullivan & Freedman, 2009). Fat soluble food are not properly digested because of failure to release bile (which aids in the digestion of fat) from the liver bile duct (Seeley & Tate, 2008). It is noted the inability to absorb fat soluble nutrients such as vitamin D leads to low bone density (Seeley & Tate, 2008).
Reproductive System
- Infertility in males.
Male semen do not contain any sperm for the reason that the ductus deferens fails to develop (O'Sullivan & Freedman, 2009; Seeley & Tate, 2008).
Cause
Cystic fibrosis is caused by a mutation in a gene that encodes for a dysfunction cystic fibrosis transmembrane conductance regulator (CFTR) protein. This gene is found on chromosome 7 and it is expressed in many epithelial cells and blood cells (Pierce, 2012). Researchers exhibits mice without the CFTR protein suffered from severe intestinal disease and pulmonary disease similar to cystic fibrosis (Clarke et al., 1994). There are reports of over 1,900 CFTR mutations in which the most common mutation is ΔF508 (Cystic Fibrosis Canada, 2012; O'Sullivan & Freedman, 2009). ΔF508 indicates a deletion of phenylalanine at position 508 (O'Sullivan & Freedman, 2009). This particular mutation accounts for 70% of North Americans and North Europeans (Davis, 2006). In order for a person to show symptoms of cystic fibrosis the person must obtain two copies of the mutated allele. Hence cystic fibrosis is considered as an autosomal recessive genetic disease (Pierce, 2012).
The CFTR gene has a length of 250kB and the protein contains, 1,480 amino acids (Davis et al., 1996). The CFTR protein function as a chloride channel with many regulatory roles including inhibition of sodium transport and calcium-activated chloride channels or regulation of ATP channels, intracellular vesicle transport, and outwardly rectifying chloride channel (O'Sullivan & Freedman, 2009). This protein is also involved in acidification of intracellular organelles by bicarbonate chloride exchange (O’Sullivan & Freedman, 2009).
Cystic fibrosis transmembrane conductance regulator (Massiah et al., 1999).

Pathophysiology
Genetics
Mutations in gene encoding the CFTR protein are classified into 6 classes. The earlier classes tend to result in a more severe illness.
- Class I: disruption in the gene transcription thus the protein is not formed
- Class II: disruption in the protein processing and thus hindering the protein from reaching its cellular location
- Class III: reduced channel sensitivity to triggers such as ATP
- Class IV: reduced duration of channel opening and ion flow through the channel
- Class V: reduced mRNA stability leading to a reduction in the number of proteins
- Class VI: reduced stability and increased turnover of the mature protein
(Ratjen, 2009)
Hypotheses:
- Direct relationship between CFTR and mucus obstruction: Abnormal CFTR leads to secretion of abnormal adhesive mucus. This hypothesis is unconfirmed.
- Indirect relationship between CFTR and mucus secretion. Dehydration Hypothesis: Disturbance in water and electrolyte concentration across epithelium. Dysfunctional CFTR cannot pump chloride ions into the external lumen and cannot regulate the rate of sodium ion reabsorption. This leads to salt buildup and subsequent water withdrawal down the osmotic gradient. These events result in water depletion which causes the airway surface liquid layer to become dehydrated and viscous (Kreda et al, 2012).
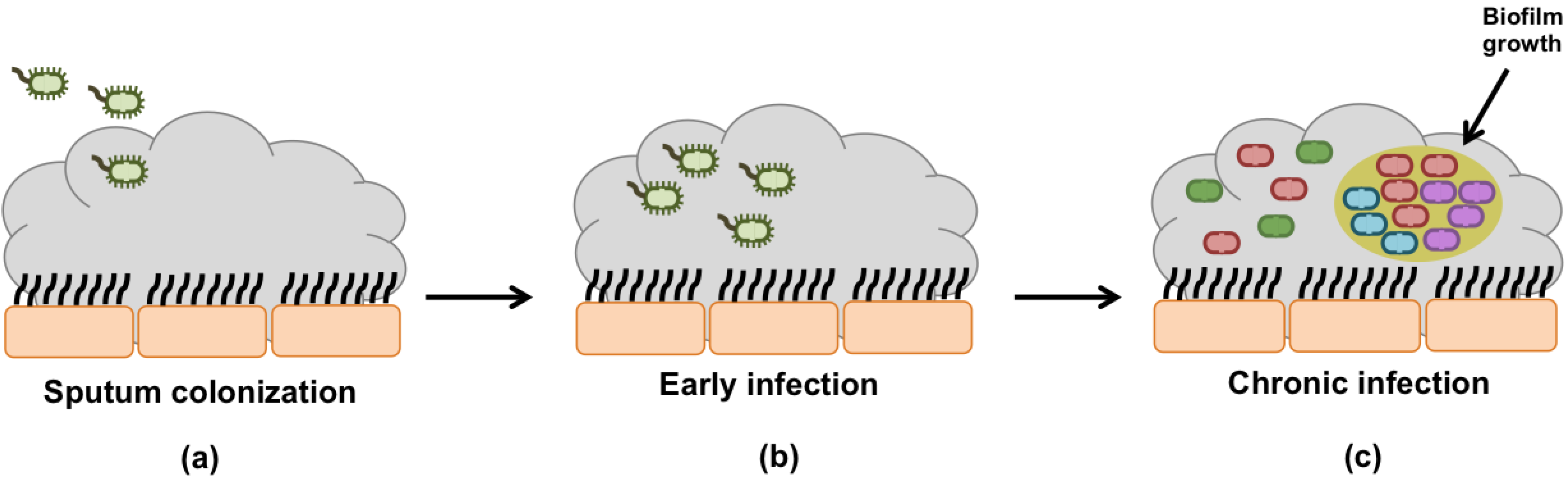
Bacterial Infection of the Airways:
The dehydrated mucus in the airway tracts becomes much harder to sweep by the cilia leading to a mucus buildup and a high susceptibility to infection. Pseudomonas Aeruginosa is the most common bacterial infection in Cystic Fibrosis. The mucus becomes hypoxic due to the high oxygen consumption by the epithelial cells. This low oxygen concentration induces changes to the Pseudomonas Aeruginosa causing it to lose motility and form aggregates that become almost impossible to eradicate (Kreda et al, 2012).
A cartoon of a bacterial infection due to cystic fibrosis (Sousa and Periera, 2014).
Diagnosis
71% of patients diagnosed with cystic fibrosis are under the age of one and 8% of patients are over the age of ten (Rosenstein & Cutting, 1998). The vast majority of cystic fibrosis patients is diagnosed through classic sign and symptoms and corroborative laboratory results (Farrell et al., 2008). The diagnosis of cystic fibrosis is based on the presence of 1 or more characteristic clinical feature, laboratory evidence of an abnormality for the CFTR gene or protein and family history or a positive newborn screening test (Rosenstein & Cutting, 1998). After a positive diagnosis for cystic fibrosis imaging techniques such as X-Rays or ultrasounds are used to evaluate the severity of the condition (Davis, 2006).
Sweat test
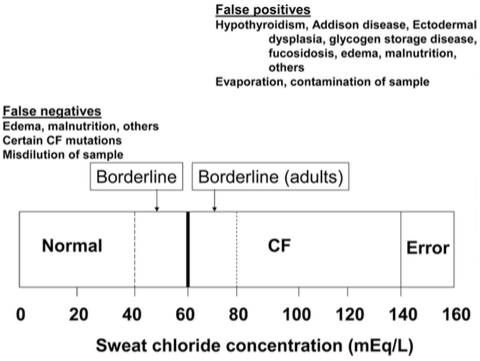
The sweat test is the primary diagnostic test for cystic fibrosis (Rosenstein & Cutting, 1998). A positive test is an observation of the sweat chloride concentration to be greater than 60 mmol/L (Davis & DI Sant’Agnese, 1984). A second positive sweat test is needed to confirm this diagnosis. It should be noted there are possibilities of a false negative sweat test since some for patients with cystic fibrosis (Davis, 2006). Patients with the false sweat test should undergo more testing such as a sperm count, assessment of the liver and gallbladder function, evidence of intestinal obstruction, and measurement of nasal potential difference (Stern et al., 1982)
Sweat chloride concentrations related to cystic fibrosis diagnosis (Davis, 2006).
Newborn Screening
Newborn screening is a laboratory technique that takes the blood of a newborn, usually about 1 week after birth (Farrell et al., 2008).Newborn screening detects high immunoreactive trypsinogen (IRT) levels (Davis, 2006). Although most patients with cystic fibrosis have elevated levels of ITR, the cutoff level capture nearly all patients with cystic fibrosis and sometimes babies without cystic fibrosis (Davidson et al., 1984) Therefore a second ITR test is followed and a sweat testing is needed (Davis, 2006).
Newborn Screening related to cystic fibrosis diagnosis (Cystic Fibrosis Foundataion, 2014).

Genetic testing
A DNA analysis is one of the methods to diagnose a person with cystic fibrosis (Rosenstein & Cutting, 1998). Commercially available genetic testing provides information about the codon region of the gene and intron sequences (Farrell et al., 2008). Often expecting parents are tested for a known cystic fibrosis genetic mutation because of a 1 in 4 chance of having a child with cystic fibrosis for heterozygous parents (Davis, 2006; Pierce, 2012). The Prenatal diagnosis tests are invasive and are accomplished by Chorionic villus sampling (CVS) and amniocentesis (Rosenstein & Cutting, 1998). These tests retrieve fetal DNA to determine its genotype (Davis, 2006). The following list is a criteria of cystic fibrosis mutations:
- Cause a change in amino acid sequence that affects the CFTR protein synthesis or function
- Introduce an early terminal signal
- Alter nucleotides of intron splice site
- Cause a new amino acid sequence that does not normally occur from at least 100 carries of the cystic fibrosis mutation from the patients ethnic group (Rosenstein & Cutting, 1998).
Treatment
Even though there is currently no cure for cystic fibrosis, many different treatment methods are being used to help monitor and ease symptoms. The main goals of these treatments include:
- Preventing and controlling lung infections
- Loosening and removing mucus from lungs
- Preventing and treating intestinal blockage
- Providing adequate nutrition
Treatment for Lung Problems:
Chest Physical Therapy:
CPT is called chest clapping or percussion and involves pounding your chest and back with your hands or a device (National Heart, Lung and Blood Institute, 2013). This process helps loosen the mucus from the lungs and allow for it to be coughed up. Sometimes sitting down or lying on ones stomach allows for better results due to the effects of gravity. Many who use CPT find it difficult to use their hands and tend to use an assisting device (National Heart, Lung and Blood Institute, 2013). These can involve an electric chest clapper, a wearable mask that creates vibrations to help break mucus and an inflatable vest that uses high frequency waves to force mucus in the lungs upwards (National Heart, Lung and Blood Institute, 2013). Along with clapping, breathing techniques with alternations between deep and relaxed breathes help loosen mucus.
Exercise:
Aerobic exercise can also help one loosen mucus so that it can be coughed out from the lungs (Mayo Clinic, 2012). The usage of exercise results in heavy breathing with helps improve your overall condition and assists in loosening mucus in the lungs. However, if individuals who have CF are working out, they should consult a doctor beforehand (Mayo Clinic, 2012). Reason being is that individuals with CF sweat with large amounts of salt. Therefore, a doctor may recommend a high-salt diet or salt supplements alongside working out to balance homeostatic salt levels in the body.
Medications:
There are many various different types of medicine that one with CF can take to help with one or many of their experienced symptoms:
- Antibiotics – used to treat and prevent lung infections that arise
- Anti-inflammatory – helps to reduce the swelling in the airways during infection
- Mucus-thinning drugs – allows individuals to cough up mucus to improve functioning of the lungs and allow proper breathing
- Bronchodilators – helps relax the muscles around the bronchiole tubules and keep the airways open
Treatment for Advanced Lung Disease:
Some individuals with CF have severe advanced lung disease. In this case, one may need oxygen therapy where oxygen is given through a nasal mask (Mayo Clinic, 2012). If this treatment fails to work, a lung transplant may be necessary where a diseased lung is replaced with a healthy lung from a donor.
Treatment for Digestive Problems:
Individuals with CF may experience many digestive problems that can lead to poor growth and development in children. As a result, some may see a nutritionist to help guide individuals with CF to a healthier lifestyle through the following:
- Oral Pancreatic Enzymes – to help digest fats and proteins and absorb vitamins
- Vitamin A, D, E and K supplements to replace the fat-soluble vitamins that are the intestines are incapable of absorbing
- High-calorie shakes with extra nutrients
- A high-salt diet or salt supplements before exercising
- Feeding tube to give one more calories while sleeping at night
Gene Therapy
Cystic Fibrosis is caused by mutation in a single-gene, the Cystic Fibrosis Transmembrane Regulator (CFTR) and approximately more than 900 mutations of this gene have been identified (Learning About Cystic Fibrosis, 2013). The Cystic Fibrosis Transmembrane Regulator basically allows proper flow of chloride and other ions from the cells. If this Cystic Fibrosis Transmembrane Regular protein structure is changed, then this results in an improper salt balance in the cells creating thick mucus (Learning About Cystic Fibrosis, 2013). Currently, studies are being conducted on finding ways to correct the defective protein.
Currently, researchers have been recommending Cystic Fibrosis Patients to undergo Gene Therapy as offers a life-saving treatment. Gene therapy basically tries to target the cause of the disease as compared to alleviating the symptoms of the disease (Learning About Cystic Fibrosis, 2013). Gene Therapy was first looked into in 1990 when researchers added normal copies of the Cystic Fibrosis Transmembrane Regulator gene to the laboratory cell cultures and successfully corrected faulty Cystic Fibrosis Transmembrane Regulator genes (Learning About Cystic Fibrosis, 2013). After this, scientists created a 'vector' by modifying a virus which was used to carry the normal genes to the Cystic Fibrosis Transmembrane Regulator cells to the affected area of the body.
The first gene therapy treatment was given to a patient in 1993 and since then, the gene therapy treatment has come a long way and several improvements have been added. Many different methods have been developed for proper gene delivery such as using nose drops, fat capsules or drizzling cells down a flexible tube to the targeted areas of the body (Learning About Cystic Fibrosis, 2013). Gene therapy is still improving and currently is facing a few problems. Scientists are still finding an effective way to deliver the genes to the affected areas in the body but other than this, researchers must also determine if gene therapy is a permanent cure to Cystic Fibrosis or does the treatment have to be repeated after a specified time period. Researchers also need to identify the cells that produce the Cystic Fibrosis Transmembrane Regulator cells and how long the affected lung cells can survive. Lungs cells were used in early gene therapy treatments because lung cells are easily accessible and also, patients most commonly have lung damage (Learning About Cystic Fibrosis, 2013).
Gene Therapy using adenovirus vector (Gene Therapy, 2007).

Recently, researchers completed an entire genetic map of the Pseudomonas aeruginosa bacterium. This was accomplished at the University of Washington's Genome Center and at the PathoGenesis Corporation. The P.aeruginosa bacteria is main cause of chronic and fatal lung infections in cystic fibrosis patients (Learning About Cystic Fibrosis, 2013). Scientists are currently studying the bacteria's genome map to find treatments to infections caused by this bacteria.
Circular representation of the P. aeruginosa genome (Stover and Pham, 2010).

Gene therapy has slowly improved over the past few years developing as an effective treatment against Cystic Fibrosis (Southern, 2012).UpGvQ4phyn0?.swf
Conclusion
Cystic fibrosis is an autosomal recessive inherited disease that mainly affects respiratory, digestive and reproductive systems in the body (Cystic Fibrosis Canada, 2012). This disease is mostly prevalent in caucasian compared to any other race (Grosse,et al., 2004). This genetic disease is caused by a dysfunction of a chloride channel called cystic fibrosis transmembrane conductance regulator (CFTR) protein (Davis, 2006). Cystic fibrosis symptoms such as persistent lung infections, poor growth, and elevated chloride concentration appears throughout life (O'Sullivan & Freedman, 2009). The standard diagnosis test is a chloride sweat test and treatments range from antibiotics to lung transplant depending on the severity of the condition (Cystic Fibrosis Canada, 2012; Welsh & Ramsey, 1998). Currently there is no cure for this disease but research such as gene therapy seems to be a promising treatment for the future (Davis, 2006).
References
Andersen, D. H. (1938). Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. American journal of Diseases of Children, 56(2), 344-399.
Chen, H. (2006). Atlas of Genetic Diagnosis and Counseling. Totowa NJ: Humana press.
Clarke, L. L., Grubb, B. R., Yankaskas, J. R., Cotton, C. U., McKenzie, A., & Boucher, R. C. (1994). Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in Cftr (-/-) mice. Proceedings of the National Academy of Sciences, 91(2), 479-483.
Cystic Fibrosis Canada. (2012). The Canadian Cystic Fibrosis Registry. Retrieved from http://www.cysticfibrosis.ca/wp-content/uploads/2014/03/Canadian-CF-Registry-English-FINAL-FOR-WEB1.pdf
Cystic Fibrosis Foundation. (2012). Cystic Fibrosis Foundation Patient Registry 2012 Annual Data Report. Retrieved from http://www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf
Davidson, A. G. F., Wong, L. T. K., Kirby, L. T., & Applegarth, D. A. (1984). Immunoreactive trypsin in cystic fibrosis. Journal of pediatric gastroenterology and nutrition, 3, S79-88.
Davis, P. B. (2006). Cystic fibrosis since 1938. American journal of respiratory and critical care medicine, 173(5), 475-482.
Davis, P.B. Drumm M. & Konstan, M.W. (1996). Cystic Fibrosis. American Journal of Respiratory and Critical Care Medicine,154(5), 1229-1256.
DI Sant'Agnese, P., Darling, R. C., Perera, G. A., & Shea, E. (1953). Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas. Pediatrics,12, 549-563.
Farber, S. (1944). Pancreatic function and disease in early life. V. Pathologic changes associated with pancreatic insufficiency in early life. Arch Pathol, 37, 238.
Farrell, P. M., Rosenstein, B. J., White, T. B., Accurso, F. J., Castellani, C., Cutting, G. R., … & Campbell III, P. W. (2008). Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. The Journal of pediatrics, 153(2), S4-S14.
Gene Therpay. (2007). Heart. Retrieved from http://heart.bmj.com/content/94/1/89/F2.expansion
Grief, J. (2008). Medical imaging in patients with Cystic Fibrosis. Retrieved from http://www.eradimaging.com/site/article.cfm?ID=327
Grosse, S. D., Boyle, C. A., Botkin, J. R., Comeau, A. M., Kharrazi, M., Rosenfeld, M., & Wilfond, B. S. (2004). Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs.MMWR Recomm Rep, 53(RR-13),1-36.
Hodson, M. (2007). Cystic fibrosis (3rd ed.). London: Hodder Arnold.
Katkin, JP. Cystic fibrosis: Genetics and pathogenesis. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on October 29, 2014.)
Knowles, M. R., Stutts, M. J., Spock, A., Fischer, N., Gatzy, J. T., & Boucher, R. C. (1983). Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science, 221(4615), 1067-1070.
Kreda, S. M., Davis, C. W., & Rose, M. C. (2012). CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harbor perspectives in medicine, 2(9), a009589.
Learning About Cystic Fibrosis. (2013). National Human Genome Research Institute. Retrieved from http://www.genome.gov/10001213
Mall, M., & Elborn, J. (2014). Cystic fibrosis. Sheffield: European Respiratory Society.
Massiah, M. A., Ko, Y.H., Pedersen, P. L. & Mildvan, A. S. (1999). Cystic Fibrosis Transmembrane Conductance Regulator: Solution Structures of Peptides Based on the Phe508 Region, the Most Common Site of Disease-Causing ΔF508 Mutation. Biochemistry, 38 (23), 7453-7461.
Mayo Clinic. (2012). Cystic Fibrosis: Treatments and Drugs. Retrieved from http://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/basics/treatment/con-20013731
National Heart, Lung and Blood Institute. (2013). How is Cystic Fibrosis Treated? Retrieved from http://www.nhlbi.nih.gov/health/health-topics/topics/cf/treatment.html
National Heart, Lung, and Blood Institute. (2013). What Causes Cystic Fibrosis? Retrieved from http://www.nhlbi.nih.gov/health/health-topics/topics/cf/causes.html
O'Sullivan, B. P. & Freedman, S.D. (2009). Cystic fibrosis. The Lancet, 373 (9678), 1891-1904.
Pierce, B. (2012). Genetics: A conceptual approach (4th ed.). New York: W.H. Freeman.
Preedy, V. (2012). Cystic Fibrosis, Sickle Cell Disease and Phenylketonuria. In Handbook of growth and growth monitoring in health and disease (Vol. 1, pp. 2337-2348). New York, NY: Springer Science Business Media, LLC.
Quinton, P. M. (1983). Chloride impermeability in cystic fibrosis. Nature,301(5899), 421-422.
Ratjen, F. A. (2009). Cystic fibrosis: pathogenesis and future treatment strategies. Respiratory care, 54(5), 595-605.
Rosenstein, B. J., & Cutting, G. R. (1998). The diagnosis of cystic fibrosis: a consensus statement. The Journal of pediatrics, 132(4), 589-595.
Seeley, R., & Tate, P. (2008). Anatomy & physiology (8th ed.). Dubuque, IA: McGraw-Hill.
Sousa, A. M., & Pereira, M. O. (2014). Pseudomonas aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs—A Review. Pathogens,3(3), 680-703.
Southern, K. [ChangingFuturesUK's channel]. (2012, January 23). The Gene Therapy Timeline - 1995, the Cystic Fibrosis Mouse [Video File]. Retrieved from http://www.youtube.com/watch?v=UpGvQ4phyn0
Stern, R., Boat, T., & Doershuk, C. (1982). Obstructive azoospermia as a diagnostic criterion for the cystic fibrosis syndrome. The Lancet, 319(8286), 1401-1404.
Stover, C., & Pham, X. (2010). Complete Genome Sequence of Bacteria. Retrieved from http://www.nature.com/nature/journal/v406/n6799/full/406959a0.html
Welsh, M. J., & Ramsey, B. W. (1998). Research on cystic fibrosis: a journey from the heart house. American journal of respiratory and critical care medicine,157(4), S148-S154.