Table of Contents
The Structure and Mechanism of an Active, Lipid-Linked Flippase
This Wiki page is adapted from a 2015 Nature publication by Perez et al. entitled “Structure and Mechanism of an Active, Lipid-Linked Oligosaccharide Flippase”. Read the full article here: https://www.nature.com/articles/nature14953
Purpose
Flippase proteins play crucial roles in cellular processes. One such flippase is PglK, a membrane-bound ABC transporter involved in bacterial N-glycosylation. PglK flips lipid-linked oligosaccharides (LLOs) from the cytoplasm to the periplasm for their subsequent addition to membrane-bound proteins (Perez et al., 2015). Prior to this study, the mechanism by which PglK carried out LLO translocation largely evaded scientists. Consequently, researchers Perez et al. (2015) set out to elucidate its mechanism by solving the structures of three different conformations of PglK. Based on the accumulated biochemical and structural data, they proposed a flipping mechanism that was distinct from most known ABC transporters (Perez et al., 2015). These seminal findings will be discussed in greater detail in this Wikipedia page.
PglK in vitro LLO Flipping and ATPase Activity
In bacteria, PglK has been proposed to perform lipid-linked oligosaccharide (LLO) translocation by using energy derived from ATP hydrolysis. To investigate the LLO flipping and ATPase activity of PglK, Perez et al. (2015) generated proteoliposomes. Proteoliposomes are similar to vesicles, as they are comprised of a phospholipid ring, lipopolysaccharide (LPS), and membrane proteins. In order to form closed vesicles, the protein of interest is generally purified and reconstituted with membrane lipids (Marasini et al., 2017). Perez et al (2015) performed this process by incorporating purified PglK into lipid vesicles containing trisaccharide LLOs (tLLOs). Di-N-acetylbacillosamine and two N-acetylgalactosamine (GalNAc) moieties were present in this tLLO (Figure 1). To determine the rate of ATP-driven tLLO flipping, the amount of left-over tLLO molecules in the outer leaflet of proteoliposomes was quantified by radiolabelling using purified PglH, an enzyme of the C. jejuni N-glycosylation machinery. PglH can attach up to three GalNAc residues to the non-reducing end of tLLOs (Perez et al., 2015). Thus, excess UDP-GalNAc was used in order to force the addition of three GalNAc molecules to each tLLO, allowing the researchers to quantify unflipped tLLOs (Figure 2). tLLO to hexa-saccharide LLO conversion was observed by transferring the resulting oligosaccharides to fluorescently labeled acceptor peptides using glycosyltransferase PglB. SDS-PAGE analysis was subsequently used to visualize the glycosylated acceptor peptides (Perez et al., 2015). The intensity of the band at a given molecular weight was related to the quantity of unflipped tLLO (Perez et al., 2015). From this, flipping rates could be determined for PglK and various mutants of the enzyme.

Figure 1: in vitro LLO flipping assay. Proteoliposomes containing PglK and tLLO.** External addition of ATP allows for tLLO flipping. Undecaprenyl tails of tLLO are shown in purple. Red hexagons represent di-N-acetylbacillosamine; yellow squares represent N-acetylgalactosamine; and yellow squares with an asterisk represent 3H-N-acetylgalactosamine. Excess ADP results in PglK inhibition, after which proteoliposomes are incubated with PglH and 3H-UDP-GalNAc to quantitate remaining tLLO in the external leaflet.
Furthermore, PglK-catalyzed in vitro flipping of LLOs was found to be directly dependent on ATP hydrolysis. Researchers came to this conclusion by comparing ATPase activity in wild type (WT) PglK and mutant PglK (E510Q), containing an additional glutamine in the Walker-B motif. This highly conserved motif provides a carboxyl residue that coordinates and stabilizes Mg2+, a crucial cofactor in ATP hydrolysis (Rai et al., 2006). Thus, E510Q has a reduced ability to utilize ATP compared to WT PglK. Consequently, the E510Q mutant displayed a near 100-fold decrease in LLO flipping rate, strongly correlating with in vivo activity (Perez et al., 2015). ATPase activity of WT PglK in the presence of LLO also followed Michaelis-Menten kinetics, with Km value of 0.46 ± 0.08 mM. This value matches the Km of ATP-dependent tLLO flipping of 0.36 ± 0.03 mM (Perez et al., 2015). This corroborates the finding that ATP hydrolysis is necessary for LLO flipping by PglK.

Figure 2: tLLO flipping of wild-type (WT) PglK and the E510Q mutant. Proteoliposomes containing only tLLO (liposomes), constant presence of 4 mM ADP (+ADP), and absence of ATP (−ATP) act as control conditions.
Finally, Perez et al. (2015) investigated if the binding of a native substrate would stimulate ATPase activity, as observed in other ABC transporters. PglK-catalyzed ATP hydrolysis was stimulated 2.5-fold by both the native LLO substrate and tLLO in detergent and in liposomes, suggesting that the terminal GalNAc and branching glucose residues of native LLO are not relevant for ATPase stimulation or flippase activity (Perez et al., 2015). Additionally, unlike related ABC transporters, high substrate specificity is required to stimulate the ATPase activity of PglK. Commonly used drugs, such as verapamil and acriflavine, displayed no stimulating effect when used as substrates (Perez et al, 2015).
Structures of PglK in Distinct States
Researchers Perez et al. (2015) then used x-ray crystallography to solve three crystal structures of the PglK mutant E510Q at a resolution of 2.9-5.9 Å (Figure 3a). Two of the three structures exhibited inward-facing conformations and were designated as “apo-inward-1” and “apo-inward-2”. The third structure was bound to ADP and possessed an outward-facing, occluded conformation, thereby referred to as the “outward-occluded” state (Perez et al., 2015).
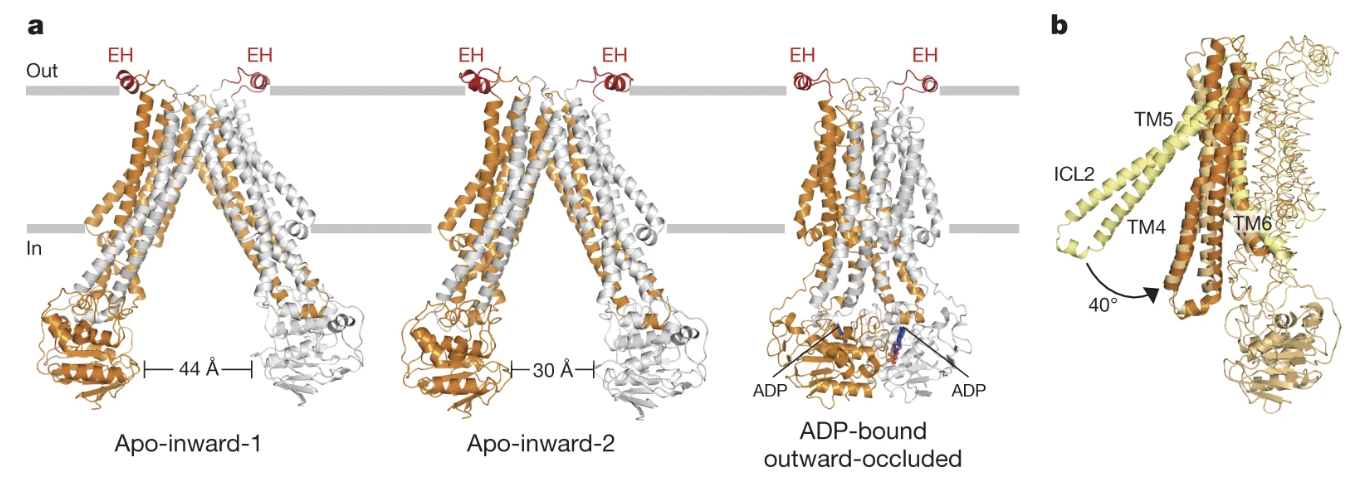
Figure 3: a) PglK is depicted as a homodimer docked in the cellular membrane. One monomer is represented in orange and the other is represented in grey. The three distinct states of PglK are shown from left to right: apo-inward-1, apo-inward-2, and ADP-bound outward-occluded. b) One monomer of PglK is shown, and transmembrane helices TM4, TM5, and TM6 are represented as ribbons. The conformational changes that result in the transition between the apo-inward-1 and outward-occluded states are also shown. TM4 and TM5 rotate a total of 40 degrees towards TM6 to transition between the two structures. The yellow helices represent TM4 and TM5 when they are in the apo-outward-1 conformation and the orange helices represent TM4 and TM5 when they are in the outward-occluded conformation.
To transition between the three distinct conformations, researchers proposed a scissor-like mechanism, whereby transmembrane helices TM4 and TM5 rotate like a rigid body towards transmembrane helix TM6 (Figure 3b). To shift from the apo-inward-1 conformation to apo-inward-2, TM4 and TM5 must rotate 16 degrees towards TM6, also bringing the nucleotide binding domains from 44Å to 30Å apart (Perez et al., 2015). To transition from the apo-inward-2 conformation to the outward-occluded state, TM4 and TM5 rotate an additional 24 degrees (Perez et al., 2015).
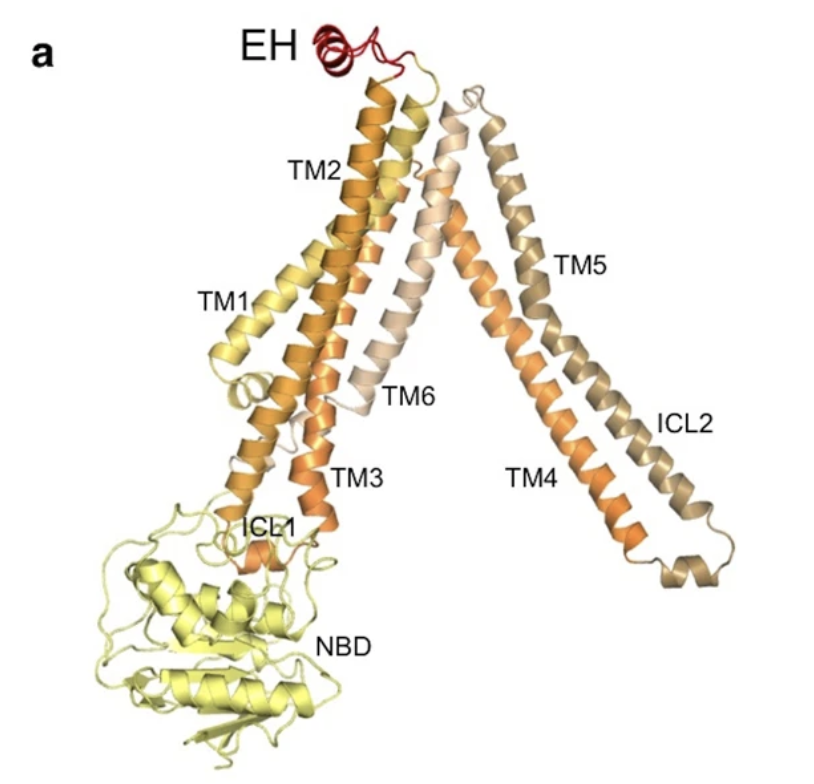
Figure 4: Structural Features of a PglK Monomer. Each structural feature is depicted in a different colour. The seven transmembrane helices are designated as TM1-TM7, the two intracellular loops are labeled as ICL1 and ICL2, the nucleotide binding domain (NBD) is shown in yellow, and the external helix (EH) is depicted in red.
Furthermore, the structure of PglK was found to be similar to that of other ABC transporters, including the first-ever determined ABC transporter, Sav1866 (Perez et al., 2015). It contains 7 transmembrane helices, designated as TM1-TM7 (Figure 4). There are two intracellular loops (IC1 and IC2) which extend into the cytoplasm of the cell, alongside a nucleotide binding domain (NBD) that is essential for ATP hydrolysis (Perez et al., 2015). Unlike Sav1866, PglK contains a short external alpha-helix, referred to as EH, that is located at the outer-leaflet of the periplasmic membrane (Perez et al., 2015). In comparing PglK to previously determined structures of ABC transporters, researchers noticed that the outward-occluded conformation of PglK has a smaller outward opening than Sav1866, yet a larger opening than an antibacterial peptide exporter McjD (Figure 5). The width of the external opening is mediated by transmembrane helices TM1 and TM3 (Perez et al., 2015). Thus, researchers concluded that this outward-occluded conformation of PglK is an intermediary state between the fully-occluded ABC transporter McjD and the fully-open Sav1866 (Perez et al., 2015).
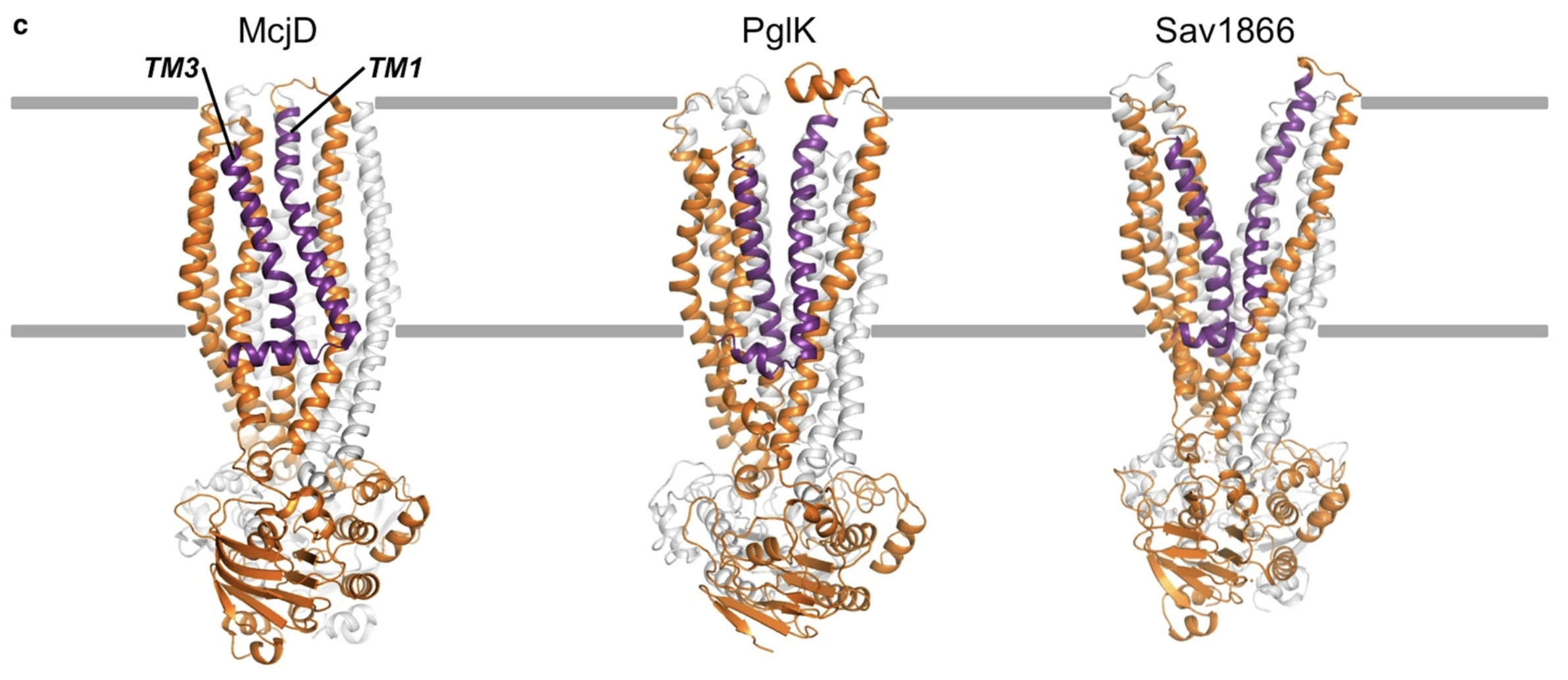
Figure 5: A Comparison of Three ABC Transporters. The bacterial ABC transporter McjD is shown on the left, PglK is represented in the middle, and Sav1866 is on the right. These three proteins possess similar structures with the exception of the two external helices unique to PglK. Additionally, PglK represents an intermediate state between the fully-occluded McjD protein and the fully-open Sav1866 transporter. The two transmembrane helices that mediate the extent of the external opening (TM1 and TM3) are shown as purple ribbons.
Role of External Helix (EH) in PglK-LLO Interaction
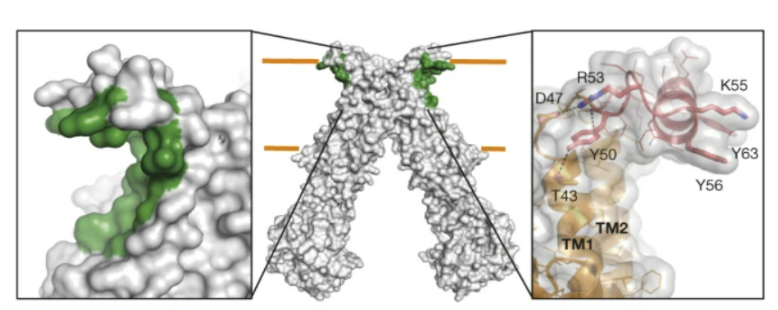
Figure 6: The EH or external helix is a part of the protein from residues S46 to P67 that is part of the loop that joins the transmembrane helices TM1 and TM2. The grooves highlighted in green are the hydrophobic grooves believed to play a role in LLO binding.
The EH or external helix is a part of the protein from residues S46 to P67 that is part of the loop that joins the transmembrane helices TM1 and TM2 (Perez et al., 2015) (Figure 6). Perez et al. (2015) generated three distinct mutants to determine how the replacement or residue mutation of the helix would affect PglK function. The deletion mutant, ΔEH, was a mutant created where a flexible linker was inserted to replace the entire EH helix. The second mutation (EHm1) involved mutating three of the tyrosine residues found in the EH into alanine residues. The third mutation (EHm2) included mutation of arginine and lysine residues involved in protein stabilization to alanines (Perez et al., 2015).
Just as in the previous component of this study, the researchers examined the wildtype versus the aforementioned mutants in liposomes and in detergent and measured ATPase activity. In the liposomes, they found that all three mutants and wildtype PglK showed similar basal ATPase activity without the presence of LLO. However, in the presence of LLO, wildtype PglK showed an increase of approximately 4 times the ATPase activity (when compared to no LLO), whilst all three mutants had no significant changes in ATPase activity (Perez et al., 2015). This suggests that the external helix is very crucial to the relationship between ATPase activity and LLO flipping. In the detergent reconstruction, in the absence of LLO, the basal level of the second and third mutants was higher compared to the wildtype, while the first mutant (linker replacement) was lower compared to the wild type (Perez et al., 2015). However, just as observed in the liposome, there was no significant increase in ATPase activity of the mutant PglK proteins when the LLO was added, confirming that the external helix is required for this event. Researchers also attempted to measure the in vitro LLO flipping rate and found that all three mutants either had significantly lowered flipping rates (15-fold and 33-fold), or could not be detected at all (Perez et al., 2015).
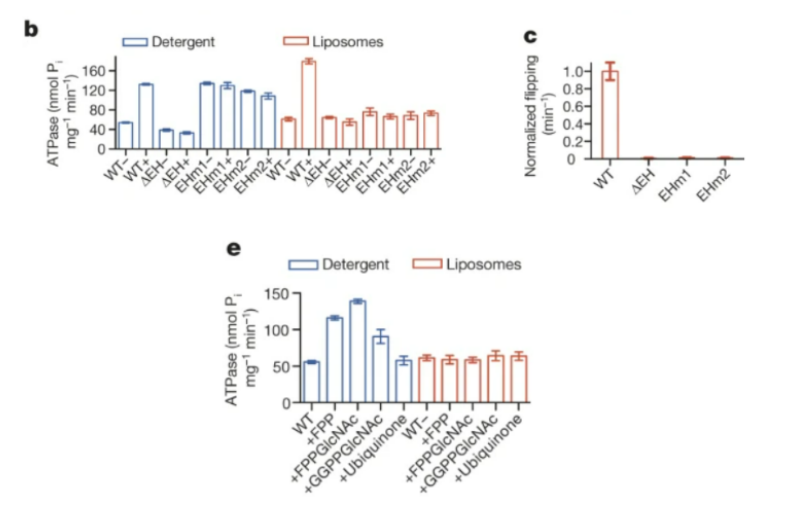
Figure 6: b) the researchers examined the wildtype versus the previously mentioned mutants in liposomes and in detergent and measured ATPase activity. c) Researchers attempted to measure the in vitro LLO flipping rate and found that all three mutants had significantly lowered flipping rates. e) Researchers used shortened but structurally similar versions of LLO to measure ATPase activity in liposomes and detergents.
The researchers also wanted to take a closer look at the role that the hydrophobic grooves found near the external helix play in LLO binding. They hypothesized that the groove may potentially interact with a LLO’s lipid tail and allowing for proper position for initiating flipping (Perez et al., 2015). It is important to note, they could not confidently confirm their hypothesis as it applied to PglK because they were not able to determine a structure for an LLO-bound PglK, and so instead they used shortened but structurally similar versions of LLO (Perez et al., 2015) . However, none of these similar structures stimulated the ATPase activity to similar levels when expressed in the liposomes. The researchers concluded that this indicates that the longer tails of the native LLO is required for recognition (Perez et al., 2015). In the detergent reconstitution, the ATPase activity was increased in the presence of several of the lipids used, however, because these results occurred in the detergent, it is possible that the increase ATPase activity is due to the binding of several molecules instead of appropriate flipping (Perez et al., 2015). Interestingly, the last structure tested, ubiquinone, which has a tail very similar to the original LLO, did not have elevated levels of ATPase activity in either detergent or liposomes, which indicated that the LLO tail is significant for PglK recognition (Perez et al., 2015).
Function of Inward and Outward Facing Cavities
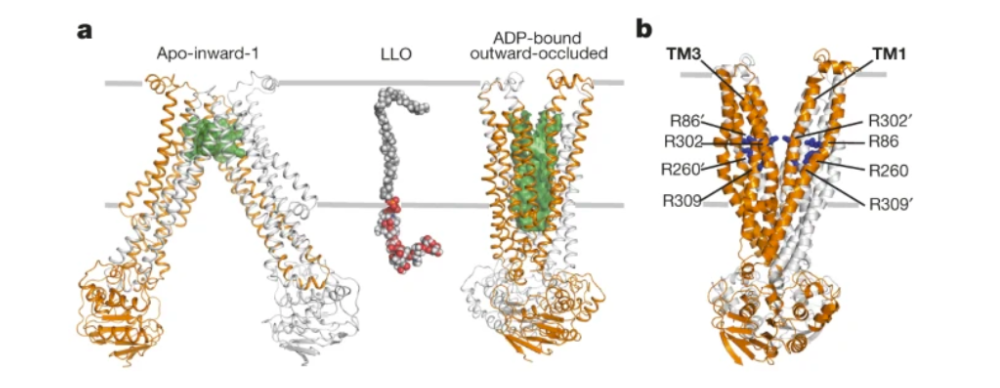
Figure 8: Inward and outward facing structures of PglK. a) PglK is shown in the Apo-inward-1 conformation on the left and the ADP-bound-outward-occluded conformation on the right. Hydrophobic cavities are represented in green. b) Arginine residues present in the outward facing PglK structure.
The inward-facing confirmation of PglK presents a large cavity towards the cytoplasmic side of the membrane. Similar cavities in other known ABC transporters function as substrate binding pockets, leading Perez et al. (2015) to hypothesize that the 55C polyprenyl tail of the LLO binds to the hydrophobic surface at the bottom of the PglK cavity during flipping seen in Figure 8a. This was tested by reducing the volume of the hydrophobic section by approximately 60% by attaching two bulky residues to each PglK subunit (S294F and V297W) (Perez et al., 2015). This mutant produced LLO flipping rates equivalent to the wildtype PglK, and ATPase activity was also unaltered (Perez et al., 2015). These results argue against binding of the polyprenyl tail and suggest that the tail is exposed to the bilayer during the flip. Perez et al. (2015) then considered the transfer of the pyrophosphate oligosaccharide head group during LLO flipping, however, they determined that this was very unlikely. This is because the TM4-TM5 areas of each PglK subunit cross over and bind the nucleotide binding domain (NBD) of the opposite subunit (Perez et al., 2015). If the LLO head group were to bind, it would be pinched off during the transition to the outward-facing state, creating a large steric clash and preventing the release of the LLO substrate (Perez et al., 2015). These results conclude that the inward-facing cavity is likely not involved in LLO binding.
The outward-facing PglK structure is distinct from the inward-facing state, revealing a wide opening to the periplasmic side of the membrane, large enough to contain the pyrophosphate and oligosaccharide components of the LLO (Perez et al., 2015). This state is also distinct in its chemical nature, possessing multiple arginine residues (R86, R260, R302, R309) that line the cavity, accounting for 8 positive charges as seen in Figure 8b (Perez et al., 2015). During the inward confirmation, these residues are buried or form salt bridges, making them accessible only in the outward confirmations (Perez et al., 2015). To determine whether these residues are involved in LLO flipping, Perez et al. introduced individual arginine mutants as well as a fourfold mutant that removed the positive charges. The individual mutants maintained a level of LLO flipping activity in vivo and in vitro as well as a lower basal rate of ATPase activity. However, the fourfold mutant was unable to catalyze LLO flipping and did not appear to have any ATPase activity, perhaps suggesting a loss of LLO and mutant PglK interaction. Thus, these arginine residues present in the outward-facing PglK structure are likely essential to the process of LLO flipping (Perez et al., 2015).
Flipping Mechanism
By utilizing their structural data, Perez et al. (2015) proposed a mechanism by which Pglk flips a LLO across the membrane (Figure 9). Three conformations exist of the protein, however, the apo-inward state was proposed to be transient and sparsely found in vivo. Therefore, only the outward-occluded (ADP-bound) and outward facing (ATP-bound) states are relevant for flipping activity (Perez et al. 2015).
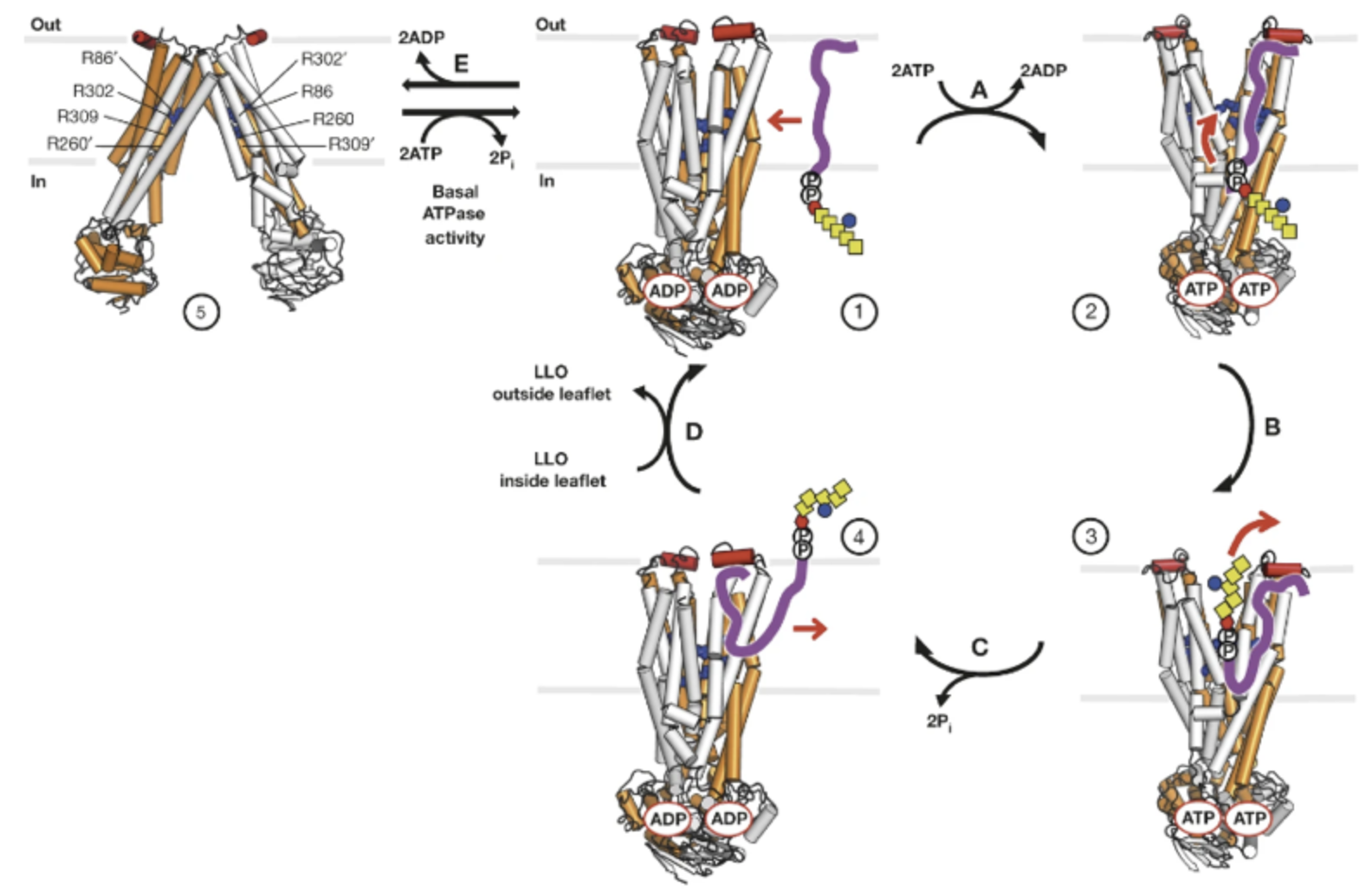
Figure 9: Proposed Flipping Mechanism of PglK. The homodimeric protein is represented as a cylinder model, with subunits shown in grey and orange. Each monomer has a binding site for ATP or ADP. PglK is situated in the membrane, between the cytoplasm (in) and periplasm (out) of a bacterial cell. The external helix is shown in red and the arginine belt shown is in blue. PglK interacts with the lipid-linked oligosaccharide (LLO), consisting of an oligosaccharide linked to a pyrophosphate (encircled P) and polyprenol tail (purple). The sugar contains one di-N-acetylbacillosamine (rex hexagon), five N-acetylgalactosamines (yellow square), and a glucose branch (blue circle). State 5 denotes the apo-inward-1 conformation, states 1 and 4 denote the outward-occluded structure, and states 2 and 3 denote outward-open conformations. Following binding, PglK flips the LLO using energy derived from ATP hydrolysis.
The flipping process begins with PglK in the outward-occluded state and the oligosaccharide of the LLO facing the cytoplasm. The polyprenyl tail of the LLO then interacts with a region on the surface of PglK that includes the external helix, leading to the exchange of bound ADP for ATP (Perez et al., 2015).. This further promotes the outward-open state of the protein. The negatively charged pyrophosphate may then enter the outward-facing cavity, where it can form electrostatic interactions with the positively charged arginine belt. The oligosaccharide linked to the pyrophosphate also enters the cavity, but does not form any specific interactions during this process. Together, the pyrophosphate and oligosaccharide make up the LLO head group (Perez et al., 2015).
ATP hydrolysis then occurs, with PglK returning to an outward-occluded state that expels the LLO to the periplasmic side of the membrane. This is achieved by two processes. Following ATP hydrolysis, the nucleotide binding domains are pushed apart, and transmission of this motion to transmembrane helices can cause pressure to be exerted on the LLO head group (Perez et al., 2015).. Also, the energy derived from ATP hydrolysis is used to break the interaction between the pyrophosphate and essential arginine residues lining the outward-facing cavity of the protein. Once the pyrophosphate and oligosaccharide have translated out of this translocation pathway, PglK adopts the outward-occluded state, thus closing the cavity to substrate binding. Finally, the polyprenol tail disengages from PglK to extend to the cytoplasmic side of the membrane, so that the LLO is facing the opposite direction as it did at the beginning of the process (Perez et al., 2015).. This mechanism resembles the ‘credit card swipe’ model utilized by P4-ATPases to translocate lipids across a membrane, albeit with two key differences. First, the polyprenol tail must be recognized by the external helix on PglK’s surface. Second, a long and wide translocation pathway is needed to contain the pyrophosphate and oligosaccharide while flipping. These requirements make PglK distinct from other flippases that engage in lipid translocation (Perez et al., 2015).
This mechanism resembles the ‘credit card swipe’ model utilized by P4-ATPases to translocate lipids across a membrane, albeit with two key differences. First, the polyprenol tail must be recognized by the external helix on PglK’s surface. Second, a long and wide translocation pathway is needed to contain the pyrophosphate and oligosaccharide while flipping. These requirements make PglK distinct from other flippases that engage in lipid translocation (Perez et al., 2015).
Methods
LLO Flipping Assay
Step 1: LLO and tLLO Extraction
To generate substrates for the flippase assays, LLOs and tLLOs were both extracted from E. coli SCM6 cells (Perez et al., 2015). LLOs were extracted from cells carrying a C. jejuni pglBmut cluster with an inactivated pglB gene, whilst tLLOs were extracted from cells with a pglHmut:pglBmut cluster with an inactivated pglH gene (Perez et al., 2015). A mixture of chloroform, water, and methanol in a ratio of 10:3:10 was used for the extraction of LLOs from the cells. Alternatively, a 1:2 ratio of methanol to chloroform was used to extract tLLOs. The extracts were then dried in a rotary evaporator (Perez et al., 2015). After drying, the extracts were reconstituted in a buffer and their concentrations were measured using titrations and an in vitro glycosylation assay (Perez et al., 2015).
Step 2: Reconstitution of PglK and Variants in Proteoliposomes
A mixture of 3:1 (w:w) of E. coli polar lipids and L-α-phosphatidylcholine liposomes were prepped to control the size and to remove any impurities by extrusion through polycarbonate filters (Perez et al., 2015). The liposomes were then saturated with Triton X-100 and mixed with purified protein. The Triton X-100 saturation allows for a better-maintained structure of the liposomes while also increasing the rate of the correct orientation of the integrated proteins (Perez et al., 2015). Biobeads were then added to remove the Triton X-100 reagent. Afterward, they were centrifuged, washed, and finally frozen in liquid nitrogen at -80 °C (Perez et al., 2015).
Step 3: Determination of PglK orientation in liposomes
Mutants of PglK that contained none of the original cysteines, but instead, had an engineered cysteine at each nucleotide-binding domain (C269L/C352S/C386S/C549L/S544C) were reconstituted in the proteoliposomes (Perez et al., 2015). The proteoliposomes were then incubated with a thiol-reactive fluorescent dye called Alexa Fluor 488 C5 maleimide. These samples were then analyzed using SDS-PAGE and a fluorescence gel scanner to calculate the ratio of PglK with NBDs facing outwards (Perez et al., 2015). This was done by comparing the band intensities obtained from non-disrupted and disrupted proteoliposomes. It was determined that 51.8 ± 2.8% of PglK molecules are orientated with the NBDs facing outwards (Perez et al., 2015).
Step 4: In vitro tLLO Flipping Assay
The PglK proteoliposomes were diluted in 10mM Tris-HCl, pH 8.0, 150 mM NaCl and 2 mM β-mercaptoethanol and extruded through polycarbonate filters with 400-nm pore size (Perez et al., 2015). They were incubated with 5 mM MgCl2 and 2 mM ATP to initiate the tLLO flipping reaction. In order to stop this reaction, the samples were diluted into a buffer containing 4 mM ADP (Perez et al., 2015). Non-flipped tLLO remnants in the external leaflet of the proteoliposomes were labeled by incubation with purified glycosyltransferase PglH (Perez et al., 2015). To stop the labeling reaction, the samples were diluted into a cold stop buffer and filtered using a Multiscreen vacuum manifold (Perez et al., 2015). Radioactivity was quantified using a gamma counter, and nonlinear fitting of data and the initial velocities were determined using GraphPad Prism 5 (Perez et al., 2015).
Step 5: Product Analysis and Determination of tLLO Orientation in Liposomes
Glycosylation of the tLLO by purified PglH was achieved through incubation of non-disrupted or disrupted proteoliposomes by adding UDP-GalNAc and PglH (Perez et al., 2015). The reaction was stopped using heat shock of 90 °C for 10 min and then the products were analyzed by in vitro glycosylation of a fluorescently labeled substrate peptide; this was catalyzed by the glycosyltransferase, PglB. The samples from this mixture were then analyzed Tricine–SDS–PAGE (Perez et al., 2015). Quantification of formed glycopeptide was determined from interpreting band intensities of the fluorescence gel scans.
Step 6: ATPase Assays
ATP hydrolysis reactions were performed using a modified molybdate-based colorimetric method (Perez et al., 2015). ATPase rates were determined using linear regression.
Step 7: Mutagenesis
PglK mutants were generated by the QuickChange method or by using gBlocks gene fragments. The plasmid constructs were then validated by DNA sequencing done by Microsynth. The PglK mutants were cloned into pMLBAD plasmids for in vivo flipping assays (Perez et al., 2015).
Step 8: Truncated LLOs Synthesis
Farnesyl- and geranyl-gernayl-PP-GlcNAc were synthesized from commercially available lipids and sugars. Since this procedure requires the formation of highly acid sensitive phosphorylated intermediates, flash column chromatography was completed on the silica gel that was basified with an ammonium hydroxide solution to prevent product degradation during the purification (Perez et al., 2015). The final LLOs were obtained from the protected precursor by deacetylation using an ammonium hydroxide solution in methanol for six hours (Perez et al., 2015). After freeze-drying, 16 mg of farnesyl-PP-GlcNAc and 42 mg of geranyl-geranyl-PP-GlcNAc were isolated as the final compounds. 1H, 13C, 31P NMR as well as negative-mode ESI-HRMS were used to show purity of the products(Perez et al., 2015).
X-Ray Crystallography
Step 1: PglK Expression and Purification
For PglK expression and purification, the gene encoding C. jejuni PglK was cloned into a modified pET-19b vector and transformed into E. coli BL21-Gold (DE3) cells for overexpression. The cells were grown at 37 °C in terrific broth (TB), and isopropyl β-d-1-thiogalactopyranoside (IPTG) was used to induce protein expression (Perez et al., 2015). Cells were then collected by centrifugation and resuspended in a solution, followed by membrane-pelleting via ultracentrifugation (Perez et al., 2015). To purify the heterogeneous mixture, a Ni-NTA affinity column and size exclusion chromatography were used (Perez et al., 2015).
Step 2: Seleno-Methionine PglK Derivative Production
Seleno-methionine PglK derivatives were used in structural determination of the outward-occluded state of the protein. To produce the derivatives, PglK was overexpressed in E. coli BL21 RIPL cells (Perez et al., 2015). These cells were grown on a culture, used to inoculate M9 media supplemented with vitamin B1 hydrochloride, and then grown in a culture supplemented with amino acids including selenomethionine (Perez et al., 2015). After 30 mins of growth with supplementation, expression was induced in the cells using IPTG. Cells were then collected (Perez et al., 2015).
Step 3: Crystallization of Native Protein
Crystallization is a critical step in structural determination via X-ray crystallography. To crystallize the outward-occluded state of PglK, the researchers incubated PglK for 0.5 hours with 5–10 mM ADP and 5–10 mM MgCl2 (Perez et al., 2015). PglK was crystallized by vapor diffusion in sitting drops and the hanging drops were crystallized at 20 degrees celsius using reservoirs (Perez et al., 2015). For the crystallization of the apo-inward-1 state, a reservoir of 100 mM Tris-HCl, pH 8.2; 150 mM Mg-acetate; 25% PEG400 was used (Perez et al., 2015). A reservoir of 50 mM glycine-NaOH, pH 9.4, 300 mM KCl, 50 mM MgCl2, and 21% PEG600 was used to crystallize hanging drops of the apo-inward-2 state (Perez et al., 2015). For the crystallization of the outward-occluded state 50 mM Na/K phosphate, pH 7.5, 250 mM NaCl, and 3.5 M ammonium sulfate was utilized as a reservoir (Perez et al., 2015). The crystals were cryoprotected by gently increasing the cryoprotectant concentration in the drops and directly flash-frozen by immersion in liquid nitrogen (Perez et al., 2015).
Step 4: Crystallization of Heavy-Metal Derivatives
To make heavy-metal derivatives, the native crystals were soaked in 1mM ethyl mercury phosphate (EMP) or 1mM potassium tetrachloroplatinate and then back soaked and flash frozen by liquid nitrogen (Perez et al., 2015).
Step 5: Data Collection
The crystals produced belong to different spatial groups. P1 was apo-inward-, P21 was apo-inward-2, and P43212 was the outward-occluded crystals. Native crystal data was collected at beamline X06SA at the Swiss Light Source (Perez et al., 2015). The 06SA (PXI) is the first macromolecular crystallography beamline at the Swiss Light Source. Next, data was processed and merged with XDS (X-ray Detector Software) and then anisotropic scaling/ellipsoid truncation was performed (Perez et al., 2015). The produced diffraction pattern was utilized to produce an electron density map. Karplus CC was then used to improve the usable resolution and quality of the electron density map (Perez et al., 2015).
Step 6: Structure Determination
Apo-Inward-1: Experimental phases of the apo-inward-1 structure were determined using multiple isomorphous replacement with anomalous scattering (MIRAS) (Perez et al., 2015). Miras is a combination of two techniques, multiple isomorphous replacement (MIR), and anomalous scattering. MIR is used to solve the phase problem in x-ray crystallography. It uses heavy derivatives of protein crystals and compares data sets of native crystals and heavy derivative crystals (Taylor, 2010). Anomalous scattering is a technique used to establish the phase of a reflection in the heavy atom derivative data (Taylor, 2010). The determination of this reflection allows researchers to establish the phase of the corresponding reflection in the native data. Furthermore, mercury positions (heavy derivative), which were used in MIR and anomalous scattering were found using SHELX and refined using SHARP (Perez et al., 2015). SHELX is a refinement program used for high-resolution diffraction data, while the SHARP program provides experimental phase information (Taylor, 2010). Solvent flattening was also done with the Solomon program. This process is performed to remove solvent peaks and valleys of densities which may appear in an electron density map due to poor quality. The densities are removed which provides a better-quality map of the protein. Electron density maps were then calculated using CCP4 programs (Perez et al., 2015). Finally, the protein model was built using Coot and refined using Phenix. Phenix is a program that can be utilized to build a protein model, refine it, validate it, and rebuild it. Coot is a similar program used to build macromolecular models from x-ray data, this program allows one to manually build macromolecules (Perez et al., 2015). CCP4 programs are a collection of data and programs which can be used in structure determination.
Apo-inward-2 structure: The structure of Apo-inward-2 was solved by molecular replacement using a partial model from the apo-inward-1 structure in Phaser (Perez et al., 2015). Phaser allows researchers to perform molecular replacement and experimental phasing methods for phasing crystal structures of macromolecules. Refinement was then performed using Phenix combined with manual building in Coot (Perez et al., 2015).
Outward-occluded state: The structure of the outward-occluded state was solved by molecular replacement using a partial model from the apo-inward-1 structure, and derivative data. A single wavelength anomalous dispersion (SAD) data set from a selenomethionine derivative was utilized (Perez et al., 2015). SAD is a technique that uses a single dataset at a single appropriate wavelength (Taylor, 2010). It is a method to determine the diffraction phases in protein crystallography. Data was also used from a derivative that had its cysteine and histidine residues labeled with EMP and PtCl4, respectively, in order to confirm the polypeptide sequence (Perez et al., 2015).
Current Scope of PglK Research
Perez et al 2015: Structure and Mechanism of an Active Lipid-Linked Oligosaccharide Flippase
The paper outlined the structure and mechanistic basis of the PglK flippase. Notably, it revealed that PglK-catalyzed LLO flipping is dependent on ATP hydrolysis. Further, PglK performs LLO flipping in the outward-facing state (Perez et al., 2015). The findings also showed that the pyrophosphate-oligosaccharide head group of the LLO enters the translocation cavity, interacting with a cluster of positively charged arginine residues. The polyprenyl tail was also found to bind to the external helix of the protein, stimulating flipping activity. Although this paper provide an in-depth exploration of PglK, further research needs to be conducted in order to fully understand this protein (Perez et al., 2015).
Perez et al 2017: Structural Basis of Inhibition of Lipid-Linked Oligosaccharide Flippase PglK by a Conformational Nanobody
The 2015 study showed PglK has two apo-inward states with large separations of the NBDs, as well as an occluded state, containing an outward facing translocation cavity. In order to investigate additional states, researchers studied the effects of camelid nanobodies (Nb) on PglK activity (Perez et al., 2017). These nanobodies facilitate protein crystallization, as they preferentially bind concave surfaces including protein active sites. Analyses showed that one inhibitory Nb binds as a single copy to homodimeric PglK. The co-crystal structure of Nb-bound PglK revealed a novel, inward-open conformation. This inhibitory state suggests tight coupling between ATPase sites, which may be applicable to other ABC transporters (Perez et al., 2017).
Perez et al 2019: Structure of Outward-Facing PglK and Molecular Dynamics of Lipid-Linked Oligosaccharide Recognition and Translocation
The previous research proposed a LLO translocation mechanisms, where the outward-facing conformation played an essential role. To investigate this postulated mechanism, researchers determined a high-resolution structure of PglK adopting an outward-occluded state with two bound ATPγS molecules (non-hydrolysable ATP analogs)(Perez et al., 2019). They then performed molecular dynamics simulations of LLO recognition and flipping based on this structure. The results suggest a probable location of an LLO recognition site on the protein surface. Further, a possible pathway for LLO flipping was revealed (Perez et al., 2019). Additional results corroborate that recognition of LLO by a cluster of positively charged arginine residues on the surfacee of PglK facilitates the specific selection for the negatively charged pyrophosphate group of LLO. Moreover, the results suggest that the ATPase site of the protein hydrolyze ATP in sequential order. This is followed by the closing of the outward-facing cavity. (Perez et al., 2019). This cavity is characterized by a vase-like shape and a narrow neck. The bottom of the cavity is predominantly negatively charged, whereas the narrow neck is positively charged, as it contains the essential “arginine belt” (Perez et al., 2019).
Unfortunately, there is a lack of research on PglK, with the papers by Perez et al providing some of the only insight into the protein’s structure and function. Despite this, growing research on other enzymes in the protein glycosylation (pgI) pathway of Campylobacter jejuni continues to be conducted.
Conclusion
This paper made seminal discoveries that revolutionized the field of membrane transporter studies. Prior to this study, little was known about the structure and mechanism of active, flippase-catalyzed lipid translocation. There was only one flippase structure solved, but no mechanism could be deduced. Thus, PglK provides novel insight into this process. There were also few in vitro assays that allowed for the study of native substrate flipping. Perez et al. (2015) generated their own flipping assay via the construction of proteoliposomes. This will allow for increased efficiency in studying flipping reactions in the future. Finally, as LLO flipping is essential for protein glycosylation, understanding its mechanism can allow novel therapeutics to be generated that can interfere with this pathway. This can decrease the virulence of many bacteria, such as the GI pathogen C. jejuni, that rely on glycosylation for their pathogenesis.
References
Marasini, N., Ghaffar, K.A., Skwarczynski, M., Toth, I. (2017) Micro and Nanotechnology in vaccine Development: Liposomes as a vaccine delivery system. Elsevier, 12, 221-239. https://www.sciencedirect.com/science/article/pii/B9780323399814000129#section-cited-by
Perez, C., Gerber, S., Boilevin, J., Bucher, M., Darbre, T., Aebi, M., Reymond, J. L., & Locher, K. P. (2015). Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature, 524(7566), 433–438. https://doi.org/10.1038/nature14953
Perez, C., Köhler, M., Janser, D. et al. (2017). Structural basis of inhibition of lipid-linked oligosaccharide flippase PglK by a conformational nanobody. Sci Rep, 7(46641). https://doi.org/10.1038/srep46641
Perez, C., Mehdipour, A., Hummer, G., Locher, K. (2019). Structure of Outward-Facing PglK and Molecular Dynamics of Lipid-Linked Oligosaccharide Recognition and Translocation, Elsevier, 27(4), 669-678. https://www.sciencedirect.com/science/article/pii/S0969212619300139
Rai, V., Gaur, M., Shukla, S., Ambudkar, S., Komath, S., Prasad, R. (2006). Conserved Asp327 of Walker B Motif in the N-Terminal Nucleotide Binding Domain (NBD-1) of Cdr1p of Candida albicans Has Acquired a New Role in ATP Hydrolysis. Biochemistry, 45(49) 14726-14739. https://pubs.acs.org/action/showCitFormats?doi=10.1021%2Fbi061535t&href=/doi/10.1021%2Fbi061535t
Taylor, G. L. (2010). Introduction to phasing. Acta Crystallographica Section D Biological Crystallography, 66(4), 325–338. https://doi.org/10.1107/s0907444910006694