Table of Contents
Structural Basis for Unidirectional Export of Lipopolysaccharide to the Cell Surface
Tristan W. Owens, Rebecca J. Taylor, Karanbir S. Pahil, Blake R. Bertani, Natividad Ruiz, Andrew C. Kruse & Daniel Kahne | Nature volume 567, pages 550–553 (2019)
The LPS Transport System in Gram-Negative Bacteria
Purpose of the Paper (Hypothesis)
Gram-negatives are inherently more resistant to antibiotics due to their outer membrane. The outer membrane is uniquely composed of lipopolysaccharides (LPS) on the outer leaflet and phospholipids on the inner leaflet. Since antibiotic resistance continues to be an issue, many researchers are interested in the outer membrane in order to develop drugs that can inhibit its synthesis and stability. The Kahne Lab is particularly focused on this, focusing their research on LPS transport. LPS is an essential component of the outer membrane. In this paper, Owens et al. sought to understand the structural components of the LPS transport system and how LPS that is synthesized in the inner membrane can be delivered onto the outer leaflet of the outer membrane. By understanding the mechanism of transport and the role of each Lpt protein, drugs can be developed to inhibit this process.
The authors used structural, biochemical, and genetic experiments to identify the path taken by LPS, from its synthesis to its entry into the cavity of the transporter to its travel up the periplasmic bridge and ultimately, to its final destination on the outer membrane. Their research also revealed which steps were ATP-dependent and how unidirectional transport of LPS was ensured. Altogether, their research has improved our understanding of LPS transport, opening doors to novel drug targets.
About the Authors
Tristan W Owens – a postdoctoral fellow at University of California. He worked under Dr. Daniel Kahne during his graduate level studies at Harvard University. His graduate research focused on LPS transport in gram-negative bacteria.
Daniel Kahne – a professor in the Department of Chemistry and Chemical Biology at Harvard University. His lab is interested in resolving antibiotic resistance by studying outer membrane components of gram-negative bacteria. Their goal is to develop strategies that can interfere with the assembly of the outer membrane.
Relevant Methods
Crystallization
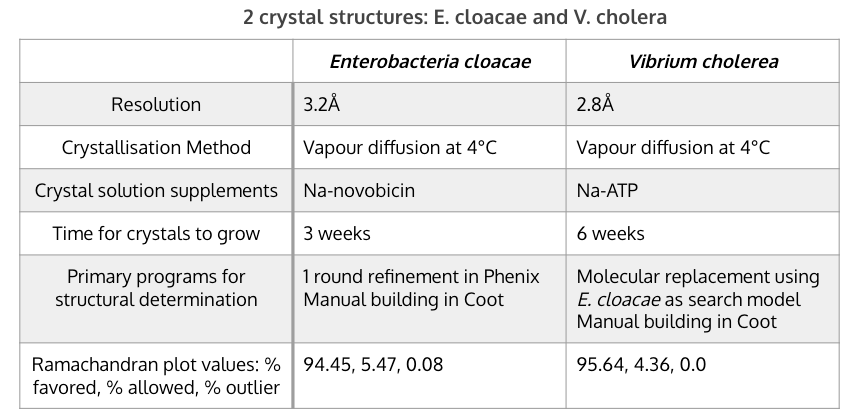
Table [1]. Key features of and differences between the two crystallisations of LptB2FGC in E. cloacae and V. cholerae.
Crystallography was used to isolate, purify, and visualize the protein structure of LptB2FGC. Two different bacterial vectors were used, Enterobacteria cloacae and Vibrium cholera. The Vibrium cholera model was slightly better, with a higher resolution and better Ramachandran values (Table 1). This may be because the Enterobacteria cloacae structure was used as a search model when building the structure for V. cholera, essentially allowing the researchers to improve upon it with V. cholerae. Na-novobiocin was added as a supplement in the crystal solution for E. cloacae and Na-ATP was provided in the solution for V. cholerae. For the former, novobiocin was required for stability and crystal contact formation, but it does not bind to a functionally significant site so it is unlikely to have altered much of importance in the structure. For V. cholera, adding ATP was not required but was kept as it drastically improved the crystal quality. The key features and differences between the crystallisations are summarized in Table 1.
The use of proteoliposomes for studying the LPS transport system
To examine the function of Lpt proteins, wild-type and variants of Lpt proteins were reconstituted into proteoliposomes. Proteoliposomes are synthetic vesicles with a lipid bilayer that closely resemble those found in bacterial cells. It provides a simplified and controlled system for studying the structure, function, interactions, and molecular mechanisms of the LPS transport system. For example, Owens et al. studied how LptB2FG complexes functioned when LptC alterations were made in proteoliposomes (Refer to Owens et al. Figure 2c). The role of LptC could then be isolated and characterized within a controlled environment. Though proteoliposomes are excellent preliminary model systems, membrane proteins should ultimately be studied in vivo under natural conditions as living cells are complex and rely on a web of interactions.
Photocrosslinking

Figure 1. Procedures for performing a photocrosslinking experiment using the amber suppression method. Figure created using BioRender. Adapted from Miyazaki et al. (2020).
Photocrosslinking was used extensively by Owens et al. to study the interactions between different Lpt proteins and between Lpt and LPS. Protein-protein interactions were confirmed by the presence of crosslinks between the structures of interest. For example, crosslinks were found between LptC and LptF, providing evidence that the two Lpt proteins may interact via β-jellyroll domains. Similarly, crosslinks between LptC and LPS were found at the base of the cavity. In general, photocrosslinking involves the incorporation of photo-activatable, unnatural amino acids (UAA) into the protein of interest (POI). This can be done using two strategies: (1) amber suppression or (2) surrogate incorporation (Mishra et al., 2019). Upon successful incorporation of the UAA, the POI is subject to ultraviolet (UV) irradiation. A covalent bond will be formed between the POI and the molecule it interacts with. Samples are then separated by size using gel electrophoresis. Western blotting is then used to confirm the presence and identity of the crosslinked proteins.
In this paper, the amber suppression method was used to incorporate p-benzoyl-L-phenylalanine (pBPA) into Lpt proteins (LptF, LptG, and LptC). pBPA is an unnatural amino acid with a benzophenone moiety that can be activated by UV light (~365 nm). Amber codons, also known as stop codons, typically signal the end of translation, but can also be used in genetic engineering to induce specific mutations (Miyazaki et al., 2020). The amber suppression method takes advantage of amber codons by replacing it with a codon that codes for an unnatural amino acid (UAA) in the POI. A novel codon will require a tRNA with a novel anticodon. A genetically engineered aminoacyl-tRNA synthetase is created to charge tRNA molecules with UAA. The UAA-charged tRNAs are then introduced into the cell or model systems. During translation, UAA will be delivered at the amber codon site (UAG). The POI will now contain the UAA at a specific site where crosslinking reactions can occur.
Western Blotting
The next step in the photocrosslinking procedure involves a Western blot, a technique used to quantify protein expression levels and to examine protein-protein interactions. Western blotting involves several steps: (1) gel electrophoresis or SDS-PAGE, (2) protein transfer to a membrane, (3) blocking the membrane to prevent non-specific binding, (4) incubation with primary antibody, (5) incubation with secondary antibody, and (6) detection of protein(s). In the first step, the protein mixture is subject to the denaturing conditions of SDS-PAGE and separated based on molecular weight. The proteins are then transferred to a polyvinylidene fluoride (PVDF) membrane. To study Lpt-Lpt protein interactions, rabbit antisera, containing primary antibodies, against LptF, LptG, and LptC are used to detect specific protein bands. The antibodies are specific to the POI and non-specific binding is washed away. Donkey anti-rabbit HRP (horseradish peroxidase) conjugate secondary antibodies are then added. To study Lpt-LPS interactions, anti-LPS core mouse monoclonal antibodies with HRP conjugates were used. The secondary antibodies are conjugated with HRP which acts as a detection molecule. ECL Prime, a chemiluminescent substrate, is used to detect protein bands. ECL Prime generates light when it reacts with HRP. Lastly, a cationic stain (Azure c600) was used to visualize proteins within the polyacrylamide gel (Owens et al., 2019). Overall, Western blotting is qualitative and only semi-quantitative. The thickness of the protein bands corresponds to the amount of protein present, but remains relative to the standard used for comparison. The absolute protein level is difficult to obtain.
The use of ADP-vanadate
Vanadate (VO4-) is a vanadium ion that binds to phosphate groups. Its binding to ADP generates ADP-vanadate which is an inhibitor of ATPases, preventing the hydrolysis of ATP. In the LPS transport system, ADP-vanadate will competitively inhibit LptB ATPase by mimicking ATP at the active site. The introduction of ADP-vanadate into the transport system allows researchers to study LPS transport in the absence of ATP without compromising other cellular processes. This was found to influence crosslinking interactions and the movement of LPS out of the cavity and onto the LptA periplasmic bridge. For example, in Figure 3d of Owens et al.’s paper, weak crosslinking of LPS to LptC with the F78 pBPA substitution was observed in the presence of vanadate.
Overview of Transport Machinery

Figure 2. Schematic Drawing of LptB2FGCADE. Created using BioRender.
 }}
}}
Figure 3. Ribbon diagram of LptB2FGC, with LptC in pink, the two copies of the ATPase LptB in brown and the transmembrane components LptF and LptG in green and blue, respectively. The membrane is shown in grey. Recent structures of LptB2FG were not able to provide insight into the pathway that lipopolysaccharides (LPS) take during their transport through the bacterial membrane. To address this, Owens et al. (2019) searched for functional protein expression in homologues of LptB2FGC from various Gram-negative bacteria. Through their investigation, they obtained crystals of LptB2FGC complexes from //Vibrio cholerae// and //Enterobacter cloacae//, which they refined to resolutions of 2.85 Å and 3.2 Å, respectively. Figure 1b from Owens et al. (2019).
The process of transporting lipopolysaccharides (LPS) across bacterial membranes is a complex and essential process for the survival of Gram-negative bacteria. LPS is a major component of the outer membrane of these bacteria, and it serves as a protective barrier against the host immune system, antibiotics, and other environmental stresses. The biosynthesis of LPS occurs in the cytoplasmic membrane and is then transported across the periplasm, a space between the cytoplasmic and the outer membrane. Once LPS biosynthesis is complete in the outer leaflet of the inner membrane, it needs to be transported to the outer membrane for assembly. This transport process is accomplished by a protein bridge composed of seven conserved LPS-transport proteins, collectively called LptB2FGCADE (Owens et al., 2019). These proteins form a channel or tunnel that spans the periplasmic space and transfers LPS from the inner membrane to the outer membrane. The LptB2FG proteins are part of the ATP-binding cassette (ABC) transporter family, which is a ubiquitous group of proteins that use the energy from ATP hydrolysis to transport various substrates across membranes. In the case of LPS transport, the energy from ATP hydrolysis is used to move LPS across the periplasmic bridge. The LptB2FG complex interacts with other components of the LPS transport machinery, including LptC, LptA, LptD, and LptE. LptC is anchored in the inner membrane by a single transmembrane helix and forms a stable sub-complex with LptB2FG. LptC receives LPS from LptF-LptG and transfers it to LptA, a soluble protein that bridges the periplasmic space. LptA interacts with the periplasmic domains of LptC and LptD, which are both integral membrane proteins, and together they form a protein bridge across the periplasm (Owens et al., 2019). LptD and LptE are part of the LptDE translocon, which forms a β-barrel structure in the outer membrane and facilitates the final insertion of LPS into the outer membrane (Owens et al., 2019). Overall, the LPS transport machinery is a highly complex and coordinated process that involves multiple components and energy sources to ensure the efficient and accurate transport of LPS from the inner membrane to the outer membrane of Gram-negative bacteria.
Their analysis revealed that LptF and LptC interact with each other to form a continuous β-jellyroll structure, which is created by the edge-to-edge interaction between their C- and N-terminal β-strands (Owens et al., 2019). Additionally, they observed multiple contacts between the side chains on the convex surfaces of the LptF and LptG β-jellyroll sheets (Owens et al., 2019). These findings provide new insights into the structural details of the LptB2FGC complex and the interaction between LptF and LptC (Owens et al., 2019).

Figure 4. Ribbon diagram depicting the view from the periplasm into the cavity between the transmembrane helices. The transmembrane anchor of LptC interdigitates between LptG transmembrane domain (TM)1 and LptF TM5 (Owens et al., 2019). To our knowledge, no other ABC system contains a transmembrane helix from another protein incorporated directly into the transporter which regulates transport activity. Figure 1c from Owens et al. (2019).
How LPS Moves Into the Cavity
Relevance of Asymmetry in Structure
In order to prove the relevance of the crystal structures, photocrosslinking between the Lpt proteins was accomplished. Owens et al. (2019) incorporated pBPA at different sites within the LptF, LptG, and LptC proteins, and subsequently tested for the presence of protein-protein interactions. A replacement was made at the interface between the LptC and LptF, and at the terminal β-strand of the LptG β-jellyroll. The results showed extensive crosslinks between the LptC and LptF proteins, while only 1 crosslink between LptC and LptG. Thus, they concluded that the asymmetry found within the crystal structure of the LPS pathway is physiologically relevant.

Figure 5. In vivo photocrosslinking of E. coli LptB2FGC complexes containing pBPA and pulling down His-tagged LptC and immunoblotting with antibodies against LptC, LptF and LptG resulted in the detection of adducts. Owens et al. (2019) Figure 2b.
Role of LptC Transmembrane Helix

Figure 6. The reconstitution of E. coli LptB2FG or LptB2FGC complexes into proteoliposomes containing LPS and their ability to transport LPS to LptA was assessed. LptC(ΔTM) represents the wild-type LptC or LptC with the transmembrane helix removed in proteoliposomes. Owens et al. (2019) Figure 2c.
An experiment involving LPS-containing proteoliposomes that harnessed E. coli LptB2FG or LptB2FGC complexes was conducted to assess their ability to transport LPS to LptA. Furthermore, this experiment sought to determine the role of the LptC transmembrane helix, as mutants with the transmembrane helix removed (LptC(ΔTM)) and mutants without LptC were explored. In the absence of LptC, LPS transport was significantly decreased, whereas the LptC(ΔTM) mutant supported LPS transport to a similar level as the wild-type LptC protein.

Figure 7. The monitoring of ATP hydrolysis by LptB2FG or LptB2FGC in proteoliposomes based on the inorganic phosphate levels measured. The three biological replicates averages are represented by bars, with error bars showing the standard deviations. Owens et al. (2019) Figure 2d.
To continue the investigation of the role of the transmembrane helix, Owens et al. (2019) monitored the ATPase activity of the LptC(ΔTM) mutant within the LPS-containing proteoliposomes. The results reflect the notion that removing the transmembrane domain of the LptC protein creates a significant increase in ATPase activity of the protein complex, making it a less efficient mechanism for LPS transport. Therefore, the main finding of this result is that the transmembrane domain of LptC allows for efficient ATP hydrolysis coupling with LPS movement across the membranes.
Cavity as LPS binding pocket
In previous studies, the cavity of LptB2FG complex showed possible accommodation of LPS but was not able to be confirmed as structures with LPS bound in the cavity was impossible to be obtained due to LPS being displaced from the cavity due to detergent molecules (Luo et al., 2017, Dong etal., 2017). Fortunately, later studies proves that the function of the cavity is evidently to bind an LPS as the structure obtained includes LPS molecules completely buried in the cavity of the LptB2FG complex (Li et al., 2019). Owens et al. (2019) specifically attempted to determine the movement of LPS into and up the transporter through in vivo photocrosslinking. This was done by trapping LPS in two regions of the transport, which are at the interface of b-jellyroll between LptC and LptF and at the base of the transporter between LptC helix, LptG TM1, and LptF TM5. As well, Owens et al. (2019) trapped LPS at the site just outside the cavity indicated by the crosslinking to LptC M19pBPA and LptF S315pBPA and another site inside the cavity defined by crosslinking to LptC G21pBPA and LptG S30pBPA. Altogether, the base crosslinking of the transporter shows how LPS enters the cavity.

Figure 8. The cryo-EM map of LptB2FG showing the surface view and cross-sectional view of the , filtered to 4.0 Å resolution. LptF in orange, LptG in light blue, two LptB subunits in green and yellow, and LPS in dark green. The respective location fo the membranes are also labelled by blue lines. The zoomed-in view of the atomic model of LPS on the right is superimposed with the cryo-EM density in grey, with the asterisk showing the position in the second heptose in the inner core, where the outer core is attached. Li et al. (2019) Figure 1d, e.
Single entry pathway for LPS
Previous structures of the LptB2FG complex reflected an alternating-access model with two potential gates for LPS entry into the cavity, represented by structurally similar entry paths. Owens et al. (2019) sought to disprove this hypothesis with their structures of the complex. Their structures show that the presence of the LptC transmembrane helix breaks the two-fold rotational pseudosymmetry of the entry paths. As a result, the points of entry differentiate, defining only a single pathway for LPS entry into the cavity. The path is located between LptG TM1 and LptF TM5, where the transmembrane domain of LptC is located. The other entry point found between LptG TM5 and LptF TM1 is blocked due to a steric interaction created by the convex surface of the continuous LptC-LptF β-jellyroll.

Figure 9. The structures of the Pseudomonas aeruginosa LptB2FG and V. cholerae LptB2FGC. On the left, showing the Pseudomonas aeruginosa LptB2FG structure obtained from Protein Data Bank (PDB) ID 5X5Y on the right, showing the V. cholerae LptB2FGC structure (right). LptG shown in blue, LptF shown in green, and LptC shown in pink with the transmembrane domains (TM) labelled. Owens et al. (2019) Figure 2e.
How LPS Moves Up Through LptC

Figure 10. In vitro photocrosslinking experiment in E.coli LptB2FGC variants with pBPA substitutions at residues M19, G21, and F78. Photocrosslinking was observed under several conditions to determine whether ATP is required for LPS entry into the cavity and LPS extraction from the inner membrane. Protein-protein interactions between LptC and LPS were quantified using a Western Blot. Figure was taken from Owens et al. (2019) Figure 3d.
An in vitro photocrosslinking experiment was performed to determine whether entry into the cavity and extraction of LPS from the inner membrane requires ATP. The unnatural amino acid pBPA was incorporated into three locations on LptC: (1) at residue M19 located just outside the cavity, (2) at residue G21 located inside the cavity, and (3) at residue F78 located on the b-jellyroll domain between LptC and LptF. The F78 location is used to pinpoint the entry point of LPS into the periplasmic bridge to suggest successful extraction from the inner membrane. The wildtype E.coli LptB2FG transport system containing modified LptC was studied under several conditions: (1) in the absence/presence of ATP, (2) in an ATP pre-hydrolysis state (+ATP, -Mg2+, +EDTA), (3) in the presence/absence of ADP-vanadate. The ATP pre-hydrolysis state occurs when ATP is present, but Mg2+ is absent which prevents ATP hydrolysis from occurring. Similarly, the ADP-vanadate condition also inhibits ATP hydrolysis. A Western blot was used to show the amount of crosslinking between LptC and LPS. As expected, in the presence of ATP, a significant amount of LPS was found near residue F78 (LptC F78 pBPA) as LPS prepares to be passed onto the LptA bridge, whereas less LptC-LPS interactions were found outside and inside the cavity. This suggests that the extraction of LPS from the inner membrane is ATP-dependent.
However, in the absence of ATP, crosslinks between LptC and LPS were found inside the cavity (at LptC G21 pBPA). This was a novel finding which suggests that LPS entry into the cavity is ATP-independent. The finding was supported by other conditions that prevented ATP hydrolysis. Crosslinking was observed in the absence of ATP, in the ATP pre-hydrolysis state, and in the presence of ADP-vanadate. Under the same conditions, crosslinking was not observed between LPS and LptC F78 pBPA. This confirms that ATP is not required to move into the cavity, but is required to exit the cavity and move into the periplasmic bridge.
Unidirectional Transport
Lpt complex structural determination

Figure 11. Two structurally distinct conformations of the LptFC interface of V. cholerae (top) and E. cloacae (bottom). Diffraction data of E. cloacae LptB2FGC crystals and V. cholerae LptB(E163Q)2FGC crystals underwent molecular replacement in Phaser. Each Lpt complex underwent rounds of refinement in Phenix and manual building in Coot. Figure was taken from Owens et al. (2019) Figure 4b.
Diffraction data for E. cloacae LptB2FGC crystals and V. cholerae LptB(E163Q)2FGC crystals were collected at the Argonne National Laboratory. The data were uploaded to the X-Ray Detector Software and scaled in the Collaborative Computational Project Number 4 software suite program AIMLESS to produce refined crystal diffraction data. To reconstruct the structure of E. cloacae LptB2FGC, molecular replacement for the E. cloacae data was performed in Phaser. Within the Phaser program, molecular replacement is accomplished using known structures that are related or homologous to the unsolved crystal structure. To solve the E. cloacae LptB2FGC structure, transmembrane domains of LptB2FG from Klebsiella pneumoniae, two copies of LptB and one copy of LptC from E. coli were used. The resulting E. cloacae structure then underwent one round of refinement in Phenix, a software package for macromolecular structure determination. Then, manual building of the transmembrane helix of LptC and the periplasmic domains of LptF and LptG were done in Coot, used for macromolecular model building, completion, and validation. After many rounds of model building and refinement in Coot and Phenix, the E. cloacae LptB2FGC structure was produced. To solve the V. cholerae LptB(E163Q)2FGC structure, the previously solved E. cloacae LptB2FGC structure was used as a search model for molecular replacement. Then, manual building of the periplasmic domains of LptF, LptG, and LptC was done in Coot to produce the final molecular structure. The results of the E. cloacae and V. cholerae structures depict two structurally distinct states: in the E. cloacae complex, the LptFC interface is open whereas in the V. cholerae complex, the LptFC interface is closed (Fig. 4b).
LptF cysteine mutants

Figure 12. Two amino acids (S157 and I234) in the V. cholerae LptF structure were replaced with cysteines to form a spontaneous disulfide bond to enforced a closed conformation. Mutations were made using site-directed mutagenesis PCR. Figure was taken from Owens et al. (2019) Figure 4c.
To determine the relationship between the LPS transport and the open conformation, a pair of cysteines capable of forming disulfide bonds were introduced to the LptF complex to trap it in a closed state (Fig. 4c). Other mutants containing single cysteine mutations were created as well. The mutant strains were grown in lysogeny broth at 37 °C with supplementation of ampicillin, IPTG, and X-Gal when needed. Owens et al. generated plasmids encoding the single cysteine mutants (LptF(S157C) and LptF(I234C)) and double cysteine mutant (LptF(S157C/I234C)) of the E. coli LptF structure using site-directed mutagenesis PCR with the pBAD18LptFG3 plasmid as a template.
To assess the viability, expression, translation, and activity of the mutants, a complementation test, Western blot, an in-vitro LPS-transport assay, and ATPase assay were performed. The complementation test was to compare a strain lacking LptF (ΔlptF) with the LptF mutants, to assess whether the mutants can restore the wild-type function of LptF (Extended data Fig. 9a). Only the double cysteine mutant could not complement with ΔlptF, meaning LptF(S157C/I234C) lost its wild-type function, and thus behaved like the deleted-LptF phenotype.

Figure 13. Table of results testing the ability of different Flag-tagged E. coli Lptf mutants to complement the ΔlptF strain to assess the viability of the mutants. Positive complementation signifies the retention of wild-type function. The double cysteine mutant (LptF(S157C/ I234C)) does not complement the ΔlptF strain as it behaves like the deleted-LptF phenotype, thus indicating a loss of function mutation. Figure was taken from Owens et al. (2019) Extended data Figure 9a.

Figure 14.Results of an anti-Flag Western blot measuring LptF-Flag (left) and LptF(S157C/I234C)-Flag (right) levels in whole cell lysates using anti-Flag monoclonal antibodies. Results show that the double cysteine mutant is the same size as the wild-type LptF-Flag. Figure was taken from Owens et al. (2019) Extended data Figure 9b.

Figure 15. Coomassie stained and purified LptB2FGC complexes including single and double cysteine mutants on SDS-PAGE. The columns are labelled as follows: (1) LptF-Flag; (2) LptF(S157C)-Flag; (3) LptF(I234C)-Flag; (4) LptF(S157C/I234C)-Flag. Figure was taken from Owens et al. (2019) Extended data Figure 9e.
A Western blot of whole-cell lysates of Flag-tagged LptF variants (LptF-Flag and LptF(S157C/I234C)-Flag) using anti-Flag monoclonal antibodies and horseradish peroxidase confirmed that the double cysteine mutant had been expressed and translated properly, for it was the same size as the wild-type LptF-Flag protein (Extended data Fig. 9b).
To confirm the correct assembly of the LptB2FGC complexes with the single and double cysteine mutants, the complexes were purified and analyzed in an SDS-PAGE (Extended data Fig. 9e). LptF-Flag, LptF(S157C)-Flag, LptF(I234C)-Flag, and LptF(S157C/I234C)-Flag all yielded the same number and size of the bands, meaning there were no assembly defects.
The LPS-transport assay tested the ability for LPS to be released to LptA(I36pBPA) from proteoliposomes containing the LptF single and double cysteine mutants within the LptB2FGC complexes (Fig. 4d). The cells that expressed the single cysteine mutants were still functional while the cells containing the double cysteine mutant were unviable such that there was no LPS-LptA adduct present. However, upon addition of DTT (dithiothreitol), a molecule that reduces disulfide bonds, the double cysteine mutant could release LPS to LptA. The reduction of the disulfide bond allowed the double cysteine mutant to regain access to an open state, and thus allowed for LPS transport to resume.

Figure 16. LPS-transport assay to LptA(I36pBPA) from proteolipsomes in absence and presence of DTT. LPS-LptA adducts were tracked for a total of 60 minutes from wild-type LptF, single cysteine mutants of LptF, and double cysteine LptF mutants. The double cysteine mutants were the only variants introduced to DTT which led to LPS transport. Figure was taken from Owens et al. (2019) Figure 4d.
To measure the relationship between ATP hydrolysis and the open/closed conformations of the complexes, ATPase activity was assayed using the EnzChek Phosphate Assay Kit which uses a spectrophotometric method (Extended data Fig. 9f). Inorganic phosphates and absorbance values were measured to calculate ATP usage by the complexes. ATP hydrolysis occurred regardless of whether the gate in LptF was open or closed since the double cysteine mutant with (open) and without (closed) DTT could still undergo ATP hydrolysis. These results are similar to the wild-type Lpt complex whereas there was no ATP hydrolysis in the catalytically dead LptB(E163Q)2FGC complex as expected. This led to the conclusion that gate opening is not directly coupled to ATP hydrolysis. Owens et al. suggested that the movement of LPS which is driven by ATP-hydrolysis pushes the gate open, and spontaneous gate closure then occurs to reduce the rate of backward flow of LPS when the gate reopens.

Figure 17. ATPase assay of E. coli LptB2FGC reconstituted into liposomes using a spectrophotometric method. Results show that ATP hydrolysis occurred regardless of open or closed conformation of LptF. Only the catalytically dead LptB(E163Q)2FGC complex showed no ATP hydrolysis. Figure was taken from Owens et al. (2019) Extended data Figure 9f.
Discussion
Full Mechanism
To sum everything up, the authors have proposed the following model for LPS transport. First, newly synthesized LPS that is present at the inner membrane binds at the entry point defined by the LptC transmembrane domain which is intercalated between LptG TM1 and LptF TM5. Next, ATP-independent entry of LPS into the cavity past the LptC transmembrane domain occurs. This is followed by ATP-dependent cavity constriction which provides the force needed to push LPS out of the membrane. The gate in the β-jellyroll of LptF improves transport efficiency by preventing backward movement of LPS into the membrane as the cavity reopens. This ensures unidirectional transport of LPS from LptC to LptA and through LptDE to the outer membrane.
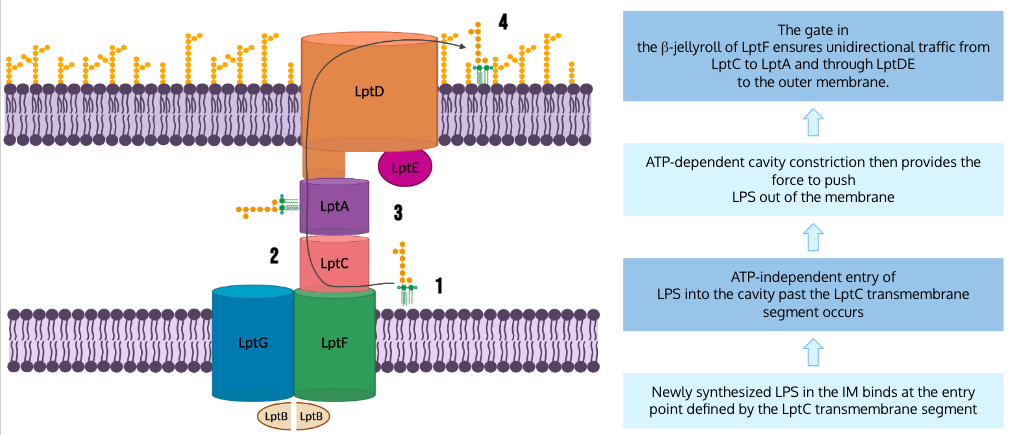
Figure 18. LPS Unidirectional Transport. This diagram depicts the full LPS transport mechanism proposed by Owens et al.. (Image created in BioRender.com).
Future Directions
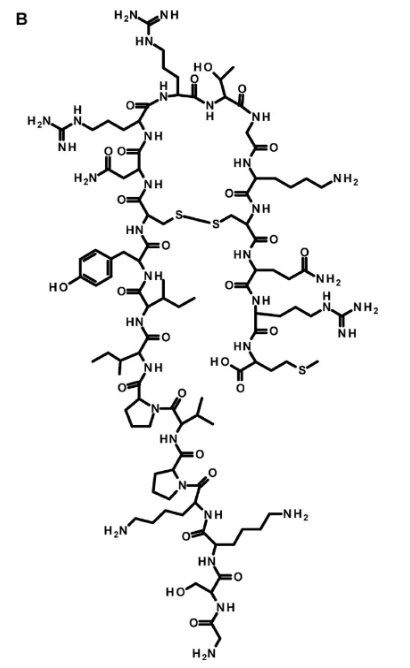
Figure 19. The structure of the novel antibiotic Thanatin. Thanatin is a pathogen-inducible single-disulfide-bond-containing β-hairpin antimicrobial peptide which was first isolated from the insect //Podisus maculiventris//. It targets LptA, which can lead to defects in the LPS transport system, and ultimately, cell death (Dash & Bhattacharjya, 2021).
LPS transport is critical for Gram-negative bacterial survival, which opens up many possibilities for the targeting of these pathways in antibiotic discovery. More specifically, there are many structures and functions within the LPS transport pathways that can act as targets, which will disrupt the unidirectional transport, and result in many adverse effects (Owens et al., 2019). For example, blocking the biosynthesis of LPS or disrupting the assembly of the nano-machinery can block the surface expression, which normally offers protection from the host immune response. Without the outer layer of LPS on the outer membrane, the bacteria become vulnerable to host defenses and the immune response (Patro & Rathinavelan, 2019). Any defects in the LPS transport system will compromise LPS insertion and assembly at the outer membrane, and result in an overall modification of the cell envelope and its permeability barrier properties. This could then allow for the entry of molecules into the bacterial cell that are typically prevented from crossing the membrane. A useful application of this consequence would be to combine antibiotics that impair the barrier with antibiotics that can be harmful to inner cellular processes (Martorana et al., 2021). Another consequence of disrupting the transport pathway is the accumulation of LPS at the inner membrane outer leaflet. The Lpt protein machinery operates as a single device, and depletion of any component leads to disruption of the whole complex (Moura et al., 2020). This irregular accumulation can then lead to adverse effects. Overall, the better our understanding is of this pathway, the better we can target it for degradation.
There is already research underway on some potential drugs that can target the LPS transport system, one of which is called thanatin. Thanatin is a naturally occurring antimicrobial peptide that is so far reported to cause defects in membrane assembly. It has been demonstrated in vitro that the drug can bind to the N-terminal β-strand of LptA. It then targets both LptC–LptA and LptA–LptA interactions, with a greater inhibitory effect on the LptC–LptA, causing loss of function. This inhibition impairs the entire LPS transport complex, by specifically causing LptA stability to decrease. While more studies are necessary on the exact effects and efficacy, it has been shown that in cells treated with thanatin, LptA undergoes degradation and LPS decorated with colanic acid accumulates. This indicates that inhibition or disruption of the Lpt complex assembly is the main killing mechanism of thanatin against Gram-negative bacteria (Moura et al., 2020).
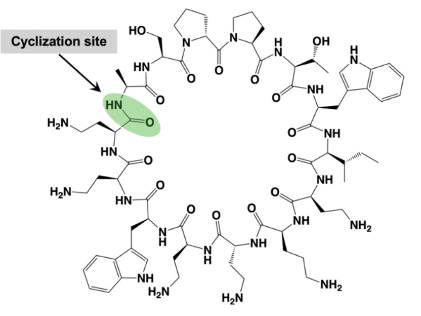
Figure 20. The structure of the novel antibiotic Murepavadin. Murepavadin, also known as POL7080, is a Pseudomonas specific peptidomimetic antibiotic. This drug is a synthetic cyclic β-hairpin host defense peptide mimetic that targets the outer membrane protein transporter LptD of the Gram-negative bacterium P. aeruginosa. This targeting can also lead to LPS transport dysfunction and cell death (Amponnawarat et al., 2021). .
Another more well studied antibiotic that targets LPS transport machinery is Murepavadin, a pathogen specific antimicrobial peptidomimetic with a novel, non-lytic mechanism of action (Martin-Loeches et al., 2018). This drug targets Pseudomonas aeruginosa, the key pathogen in cystic fibrosis (CF) lung infections (Eckford, 2023). With the current increase in multi-drug resistant P. aeruginosa, antibiotics with a novel mechanism of action are needed to target these infections (Martin-Loeches et al., 2018). Murepavadin was the first antibiotic to specifically target an essential outer membrane protein (Martin-Loeches et al., 2018). This drug binds to the periplasmic jellyroll domain of LptD of Pseudomonas, which significantly differs from LptD of other Gram-negative bacteria, blocking translocation of LPS from the periplasm to the outer membrane (Srinivas et al., 2010). This pathway also results in modified LPS accumulation, altered outer membrane LPS expression, and ultimately, cell death (Martin-Loeches et al., 2018).
These two examples prove that the LPS transport system and its function are valuable targets for antimicrobial discovery. The specificity to Gram-negative bacteria is also important, since these bacteria are known to be harder to treat than Gram-positive bacteria (Breijyeh et al., 2020). The better our understanding is of this pathway, the better we can target it. This means contributions to the literature, such as this Owens et al. paper, are crucial for future investigations.
Conclusion
The main takeaways from Owens et al’s paper are:
- The structure of the inner membrane complex of the LPS transport machinery was revealed, along with specific b-jellyroll interactions between Lpt proteins
- The transmembrane helix of LptC regulates LPS transport and improves ATP hydrolysis
- LptC creates a single entry pathway for LPS to move onto the periplasmic bridge
- Entry into the cavity is ATP-independent, whereas movement up the periplasmic bridge is ATP-dependent
- Gate in the b-jellyroll of LptF prevents backward flow of LPS to ensure unidirectional transport from LptC to LptA and ultimately, to the outer membrane
References
Breijyeh, Z., Jubeh, B., & Karaman, R. (2020). Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules, 25(6), 1340. https://doi.org/10.3390/molecules25061340
Chaudhuri, D., Ganesan, R., Vogelaar, A., Dughbaj, M. A., Beringer, P. M., & Camarero, J. A. (2021). Chemical synthesis of a potent antimicrobial peptide murepavadin using a tandem native chemical ligation/desulfurization reaction. The Journal of Organic Chemistry, 86(21), 15242–15246. https://doi.org/10.1021/acs.joc.1c01858
Dong, H., Zhang, Z., Tang, X. et al. (2017). Structural and functional insights into the lipopolysaccharide ABC transporter LptB2FG. Nat Commun 8, 222 . https://doi.org/10.1038/s41467-017-00273-5
Eckford, C. (2023). Promising first-in-human study results for orally inhaled antibiotic. European Pharmaceutical Review. https://www.europeanpharmaceuticalreview.com/news/178380/promising-first-in-human-study-results-for-orally-inhaled-antibiotic-macrocycle-compound/
Li, Y., Orlando, B.J. & Liao, M. (2019). Structural basis of lipopolysaccharide extraction by the LptB2FGC complex. Nature 567, 486–490 . https://doi.org/10.1038/s41586-019-1025-6
Luo, Q., Yang, X., Yu, S. et al. (2017). Structural basis for lipopolysaccharide extraction by ABC transporter LptB2FG. Nat Struct Mol Biol 24, 469–474. https://doi.org/10.1038/nsmb.3399
Martin-Loeches, I., Dale, G. E., & Torres, A. (2018). Murepavadin: A new antibiotic class in the pipeline. Expert Review of Anti-Infective Therapy, 16(4), 259–268. https://doi.org/10.1080/14787210.2018.1441024
Martorana, A. M., Moura, E. C. C. M., Sperandeo, P., Di Vincenzo, F., Liang, X., Toone, E., Zhou, P., & Polissi, A. (2021). Degradation of components of the lpt transenvelope machinery reveals lps-dependent lpt complex stability in escherichia coli. Frontiers in Molecular Biosciences, 8. https://www.frontiersin.org/articles/10.3389/fmolb.2021.758228
Mishra, P. K., Yoo, C. M., Hong, E., & Rhee, H. W. (2020). Photo‐crosslinking: An emerging chemical tool for investigating molecular networks in live cells. ChemBioChem, 21(7), 924–932. https://doi.org/10.1002/cbic.201900600
Miyazaki, R., Akiyama, Y., & Mori, H. (2020). A photo-cross-linking approach to monitor protein dynamics in living cells. Biochimica Et Biophysica Acta (BBA) - General Subjects, 1864(2), 129317. https://doi.org/10.1016/j.bbagen.2019.03.003
Moura, E. C. C. M., Baeta, T., Romanelli, A., Laguri, C., Martorana, A. M., Erba, E., Simorre, J.-P., Sperandeo, P., & Polissi, A. (2020). Thanatin impairs lipopolysaccharide transport complex assembly by targeting lptc–lpta interaction and decreasing lpta stability. Frontiers in Microbiology, 11, 909. https://doi.org/10.3389/fmicb.2020.00909
Owens, T. W., Taylor, R. J., Pahil, K. S., Bertani, B. R., Ruiz, N., Kruse, A. C., & Kahne, D. (2019). Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature, 567(7749), 550–553. https://doi.org/10.1038/s41586-019-1039-0
Patro, L. P. P., & Rathinavelan, T. (2019). Targeting the sugary armor of klebsiella species. Frontiers in Cellular and Infection Microbiology, 9, 367. https://doi.org/10.3389/fcimb.2019.00367
Srinivas, N., Jetter, P., Ueberbacher, B. J., Werneburg, M., Zerbe, K., Steinmann, J., Van Der Meijden, B., Bernardini, F., Lederer, A., Dias, R. L. A., Misson, P. E., Henze, H., Zumbrunn, J., Gombert, F. O., Obrecht, D., Hunziker, P., Schauer, S., Ziegler, U., Käch, A., … Robinson, J. A. (2010). Peptidomimetic antibiotics target outer-membrane biogenesis in pseudomonas aeruginosa. Science, 327(5968), 1010–1013. https://doi.org/10.1126/science.1182749
Upert, G., Luther, A., Obrecht, D., & Ermert, P. (2021). Emerging peptide antibiotics with therapeutic potential. Medicine in Drug Discovery, 9, 100078. https://doi.org/10.1016/j.medidd.2020.100078